Omenn Syndrome

Omenn syndrome is an autosomal recessive severe combined immunodeficiency. It is associated with hypomorphic missense mutations in immunologically relevant genes of T-cells (and B-cells) such as recombination activating genes (RAG1 and RAG2), Interleukin-7 receptor-α (IL7Rα), DCLRE1C-Artemis, RMRP-CHH, DNA-Ligase IV, common gamma chain, WHN-FOXN1, ZAP-70 and complete DiGeorge syndrome. It is fatal without treatment.
Symptoms
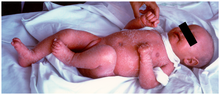
The symptoms are very similar to graft-versus-host disease (GVHD). This is because the patients have some T cells with limited levels of recombination with the mutant RAG genes. These T cells are abnormal and have a very specific affinity for self antigens found in the thymus and in the periphery. Therefore, these T cells are auto-reactive and cause the GVHD phenotype.
A characteristic symptom is chronic inflammation of the skin, which appears as a red rash (early onset erythroderma). Other symptoms include eosinophilia, failure to thrive, swollen lymph nodes, swollen spleen, diarrhea, enlarged liver, low immunoglobulin levels (except immunoglobulin E, which is elevated), low T cell levels, and no B cells.
Genetics
Omenn syndrome is caused by a partial loss of RAG gene function and leads to symptoms similar to severe combined immunodeficiency syndrome, including opportunistic infections. The RAG genes are essential for gene recombination in the T-cell receptor and B-cell receptor, and loss of this ability means that the immune system has difficulty recognizing specific pathogens. Omenn Syndrome is characterised by the loss of T-cell function, leading to engraftment of maternal lymphocytes in the foetus and the co-existence of clonally expanded autologous and transplacental-acquired maternal lymphocytes. Omenn syndrome can occasionally be caused in other recombination genes, including IL-7Rα and RMRP.
Diagnosis
In order to diagnose a patient specifically with Omenn Syndrome, an autosomal recessive form of SCID, a physician can order a genetic testing panel to look for 22q11 microdeletions or mutations of the RAG1/RAG2 genes.
Treatment
The only treatment for Omenn syndrome is chemotherapy followed by a bone marrow transplantation. Without treatment, it is rapidly fatal in infancy.
See also
- Purine nucleoside phosphorylase deficiency
- List of cutaneous conditions