Mitochondrial Disease

Mitochondrial diseases are a group of disorders caused by dysfunctional mitochondria, the organelles that generate energy for the cell. Mitochondria are found in every cell of the human body except red blood cells, and convert the energy of food molecules into the ATP that powers most cell functions.
Mitochondrial diseases take on unique characteristics both because of the way the diseases are often inherited and because mitochondria are so critical to cell function. A subclass of these diseases that have neuromuscular symptoms are sometimes called mitochondrial myopathies.
Signs and symptoms
Symptoms include:
- poor growth
- loss of muscle coordination
- muscle weakness
- visual problems
- hearing problems
- learning disabilities
- heart disease
- liver disease
- kidney disease
- gastrointestinal disorders
- respiratory disorders
- neurological problems
- autonomic dysfunction
- dementia
Acquired conditions in which mitochondrial dysfunction has been involved are:
- diabetes
- Huntington's disease
- cancer
- Alzheimer's disease
- Parkinson's disease
- bipolar disorder, schizophrenia, aging and senescence, anxiety disorders
- cardiovascular disease
- sarcopenia
- chronic fatigue syndrome
The body, and each mutation, is modulated by other genome variants; the mutation that in one individual may cause liver disease might in another person cause a brain disorder. The severity of the specific defect may also be great or small. Some defects include exercise intolerance. Defects often affect the operation of the mitochondria and multiple tissues more severely, leading to multi-system diseases.
As a rule, mitochondrial diseases are worse when the defective mitochondria are present in the muscles, cerebrum, or nerves, because these cells use more energy than most other cells in the body.
Although mitochondrial diseases vary greatly in presentation from person to person, several major clinical categories of these conditions have been defined, based on the most common phenotypic features, symptoms, and signs associated with the particular mutations that tend to cause them.
An outstanding question and area of research is whether ATP depletion or reactive oxygen species are in fact responsible for the observed phenotypic consequences.
Cerebellar atrophy or hypoplasia has sometimes been reported to be associated.
Causes
Mitochondrial disorders may be caused by mutations (acquired or inherited), in mitochondrial DNA (mtDNA), or in nuclear genes that code for mitochondrial components. They may also be the result of acquired mitochondrial dysfunction due to adverse effects of drugs, infections, or other environmental causes.
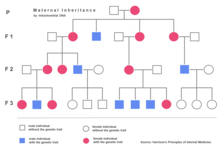
Nuclear DNA has two copies per cell (except for sperm and egg cells), one copy being inherited from the father and the other from the mother. Mitochondrial DNA, however, is inherited from the mother only (with some exceptions) and each mitochondrial organelle typically contains between 2 and 10 mtDNA copies. During cell division the mitochondria segregate randomly between the two new cells. Those mitochondria make more copies, normally reaching 500 mitochondria per cell. As mtDNA is copied when mitochondria proliferate, they can accumulate random mutations, a phenomenon called heteroplasmy. If only a few of the mtDNA copies inherited from the mother are defective, mitochondrial division may cause most of the defective copies to end up in just one of the new mitochondria (for more detailed inheritance patterns, see human mitochondrial genetics). Mitochondrial disease may become clinically apparent once the number of affected mitochondria reaches a certain level; this phenomenon is called "threshold expression".
Mitochondria possess many of the same DNA repair pathways as nuclei do—but not all of them; therefore, mutations occur more frequently in mitochondrial DNA than in nuclear DNA (see Mutation rate). This means that mitochondrial DNA disorders may occur spontaneously and relatively often. Defects in enzymes that control mitochondrial DNA replication (all of which are encoded for by genes in the nuclear DNA) may also cause mitochondrial DNA mutations.
Most mitochondrial function and biogenesis is controlled by nuclear DNA. Human mitochondrial DNA encodes 13 proteins of the respiratory chain, while most of the estimated 1,500 proteins and components targeted to mitochondria are nuclear-encoded. Defects in nuclear-encoded mitochondrial genes are associated with hundreds of clinical disease phenotypes including anemia, dementia, hypertension, lymphoma, retinopathy, seizures, and neurodevelopmental disorders.
A study by Yale University researchers (published in the February 12, 2004, issue of the New England Journal of Medicine) explored the role of mitochondria in insulin resistance among the offspring of patients with type 2 diabetes. Other studies have shown that the mechanism may involve the interruption of the mitochondrial signaling process in body cells (intramyocellular lipids). A study conducted at the Pennington Biomedical Research Center in Baton Rouge, Louisiana showed that this, in turn, partially disables the genes that produce mitochondria.
Examples
Examples of mitochondrial diseases include:
- Mitochondrial myopathy
- Diabetes mellitus and deafness (DAD)
- this combination at an early age can be due to mitochondrial disease
- Diabetes mellitus and deafness can be found together for other reasons
- Leber's hereditary optic neuropathy (LHON)
- visual loss beginning in young adulthood
- eye disorder characterized by progressive loss of central vision due to degeneration of the optic nerves and retina
- affects 1 in 50,000 people in Finland
- Leigh syndrome, subacute sclerosing encephalopathy
- after normal development the disease usually begins late in the first year of life, although onset may occur in adulthood
- a rapid decline in function occurs and is marked by seizures, altered states of consciousness, dementia, ventilatory failure
- Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP)
- progressive symptoms as described in the acronym
- dementia
- Myoneurogenic gastrointestinal encephalopathy (MNGIE)
- gastrointestinal pseudo-obstruction
- neuropathy
- Myoclonic Epilepsy with Ragged Red Fibers (MERRF)
- progressive myoclonic epilepsy
- "Ragged Red Fibers" are clumps of diseased mitochondria that accumulate in the subsarcolemmal region of the muscle fiber and appear when muscle is stained with modified Gömöri trichrome stain
- short stature
- hearing loss
- lactic acidosis
- exercise intolerance
- MELAS syndrome
- Mitochondrial DNA depletion syndrome
Conditions such as Friedreich's ataxia can affect the mitochondria but are not associated with mitochondrial proteins.
Mechanisms
The effective overall energy unit for the available body energy is referred to as the daily glycogen generation capacity, and is used to compare the mitochondrial output of healthy individuals to that of afflicted or chronically glycogen-depleted individuals. This value is slow to change in a given individual, as it takes between 18 and 24 months to complete a full cycle.
The glycogen generation capacity is entirely dependent on, and determined by, the operating levels of the mitochondria in all of the cells of the human body; however, the relation between the energy generated by the mitochondria and the glycogen capacity is very loose and is mediated by many biochemical pathways. The energy output of full healthy mitochondrial function can be predicted exactly by a complicated theoretical argument, but this argument is not straightforward, as most energy is consumed by the brain and is not easily measurable.
Diagnosis
Mitochondrial diseases are usually detected by analysing muscle samples, where the presence of these organelles is higher. The most common tests for the detection of these diseases are:
- Southern blot to detect big deletions or duplications
- Polymerase chain reaction and specific mutation testing
- Sequencing
Treatments
Although research is ongoing, treatment options are currently limited; vitamins are frequently prescribed, though the evidence for their effectiveness is limited. Pyruvate has been proposed in 2007 as a treatment option. N-acetyl cysteine reverses many models of mitochondrial dysfunction. In the case of mood disorders, specifically bipolar disorder, it is hypothesized that N-acetyl-cysteine (NAC), acetyl-L-carnitine (ALCAR), S-adenosylmethionine (SAMe), coenzyme Q10 (CoQ10), alpha-lipoic acid (ALA), creatine monohydrate (CM), and melatonin could be potential treatment options.
Gene therapy prior to conception
Spindle transfer, where the nuclear DNA is transferred to another healthy egg cell leaving the defective mitochondrial DNA behind, is a potential treatment procedure that has been successfully carried out on monkeys. Using a similar pronuclear transfer technique, researchers at Newcastle University led by Douglass Turnbull successfully transplanted healthy DNA in human eggs from women with mitochondrial disease into the eggs of women donors who were unaffected. In such cases, ethical questions have been raised regarding biological motherhood, since the child receives genes and gene regulatory molecules from two different women. Using genetic engineering in attempts to produce babies free of mitochondrial disease is controversial in some circles and raises important ethical issues. A male baby was born in Mexico in 2016 from a mother with Leigh syndrome using spindle transfer.
In September 2012 a public consultation was launched in the UK to explore the ethical issues involved. Human genetic engineering was used on a small scale to allow infertile women with genetic defects in their mitochondria to have children. In June 2013, the United Kingdom government agreed to develop legislation that would legalize the 'three-person IVF' procedure as a treatment to fix or eliminate mitochondrial diseases that are passed on from mother to child. The procedure could be offered from 29 October 2015 once regulations had been established. Embryonic mitochondrial transplant and protofection have been proposed as a possible treatment for inherited mitochondrial disease, and allotopic expression of mitochondrial proteins as a radical treatment for mtDNA mutation load.
In June 2018 Australian Senate's Senate Community Affairs References Committee recommended a move towards legalising Mitochondrial replacement therapy (MRT). Research and clinical applications of MRT were overseen by laws made by federal and state governments. State laws were, for the most part, consistent with federal law. In all states, legislation prohibited the use of MRT techniques in the clinic, and except for Western Australia, research on a limited range of MRT was permissible up to day 14 of embryo development, subject to a license being granted. In 2010, the Hon. Mark Butler MP, then Federal Minister for Mental Health and Ageing, had appointed an independent committee to review the two relevant acts: the Prohibition of Human Cloning for Reproduction Act 2002 and the Research Involving Human Embryos Act 2002. The committee’s report, released in July 2011, recommended the existing legislation remain unchanged
Currently, human clinical trials are underway at GenSight Biologics (ClinicalTrials.gov # NCT02064569) and the University of Miami (ClinicalTrials.gov # NCT02161380) to examine the safety and efficacy of mitochondrial gene therapy in Leber's hereditary optic neuropathy.
Epidemiology
About 1 in 4,000 children in the United States will develop mitochondrial disease by the age of 10 years. Up to 4,000 children per year in the US are born with a type of mitochondrial disease. Because mitochondrial disorders contain many variations and subsets, some particular mitochondrial disorders are very rare.
The average number of births per year among women at risk for transmitting mtDNA disease is estimated to approximately 150 in the United Kingdom and 800 in the United States.
History
The first pathogenic mutation in mitochondrial DNA was identified in 1988; from that time to 2016, around 275 other disease-causing mutations were identified.:37
Notable cases
Notable people who suffered from mitochondrial disease include:
- Mattie Stepanek, a poet, peace advocate, and motivational speaker who suffered from dysautonomic mitochondrial myopathy, and who died at age 13.
- Rocco Baldelli, a coach and former center fielder in Major League Baseball who had to retire from active play at age 29 due to mitochondrial channelopathy.
- Charlie Gard, a British boy who suffered from mitochondrial DNA depletion syndrome; decisions about his care were taken to various law courts.