Iminoglycinuria

Iminoglycinuria, is an autosomal recessive disorder of renal tubular transport affecting reabsorption of the amino acid glycine, and the imino acids proline and hydroxyproline. This results in excess urinary excretion of all three acids (-uria denotes "in the urine").
Iminoglycinuria is a rare and complex disorder, associated with a number of genetic mutations that cause defects in both renal and intestinal transport systems of glycine and imino acids.
Imino acids typically contain an imine functional group, instead of the amino group found in amino acids. Proline is considered and usually referred to as an amino acid, but unlike others, it has a secondary amine. This feature, unique to proline, identifies proline also as an imino acid. Hydroxyproline is another imino acid, made from the naturally occurring hydroxylation of proline.
Presentation
The primary characteristic of iminoglycinuria is the presence of glycine and imino acids in the urine. Otherwise, it is thought to be a relatively benign disorder, although symptoms associated with disruptions of proline and glycine metabolism caused by malabsorption may be present with iminoglycinuria. These include encephalopathy, mental retardation, deafness, blindness, kidney stones, hypertension and gyrate atrophy.
Gyrate atrophy is an inherited degenerative disorder of the retina and choroid, sometimes accompanying the metabolic condition hyperornithinemia. The presence of gyrate atrophy with iminoglycinuria stems from a deficiency of proline in chorioretinal tissues, while processes behind hyperornithinemia disrupt the metabolic pathway from ornithine to proline, which alters the catabolism of ornithine, and also results in reduced levels of proline. Thus, gyrate atrophy can be found with either disorder, with proline deficiency as an underlying feature.
Hyperglycinuria is another disorder affecting reabsorption of glycine and imino acids, similar to iminoglycinuria and considered to be a heterozygous form. When accompanied by a specific type of kidney stone (nephrolithiasis), it is sometimes referred to as "iminoglycinuria, type II".
Genetics

Iminoglycinuria is believed to be inherited in an autosomal recessive manner. This means a defective gene responsible for the disorder is located on an autosome, and inheritance requires two copies of the defective gene—one from each parent. Parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
A non-inherited cause of excess urinary excretion of proline and glycine, similar to that found in iminoglycinuria, is quite common to newborn infants younger than 6 months. Sometimes referred to as neonatal iminoglycinuria, it is due to underdevelopment of high-affinity transport mechanisms within the renal circuit, specifically PAT2, SIT1 and SLC6A18. The condition corrects itself with age. In cases where this persists beyond childhood, however, inherited hyperglycinuria or iminoglycinuria may be suspected.
Pathophysiology
Glycine, proline and hydroxyproline share common renal tubular mechanisms of reabsorption, a function specific to the proximal tubule. Both reabsorption or absorption of glycine and imino acids takes place respectively at the proximal tubule or intestinal brush border epithelium. The more selective transport of proline and other imino acids is driven at the molecular level by a mammalian cellular transport mechanism aptly known as system IMINO.
While no single genetic mutation has been established as the cause of iminoglycinuria; several mutations, affecting transport mechanisms shared by glycine, proline and hydroxyproline, as well as those that selectively transport either glycine or imino acids, including the IMINO system, are known to be associated with the disorder. When combined, these factors will result in a variable phenotype for iminoglycinuria depending on which mutations are present. However, despite the role that intestinal malabsorption of glycine and imino acids can play in iminoglycinuria, the primary defect disrupts their renal transport and reabsorption. This is evident, as inherited iminoglycinuria can be clinically present with no intestinal involvement.
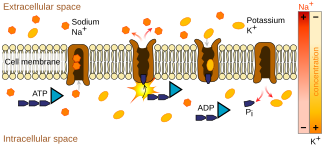
In mammals, including humans, the transport of amino and imino acids from the lumen (interior) of the intestine or the renal proximal tubule into the cells occurs at the brush border membrane of the epithelium (moist, tightly packed cellular lining of many tissues and organs of the body). Here, cotransporters such as sodium or chloride (part of the system of Na-K-Cl cotransporters) couple with the amino or imino acids on the molecular level and transport them through specific integral membrane proteins that form ion channels, which are located within the cell membrane. From the cells, the absorbed or reabsorbed amino and imino acids eventually reach the blood. Absorption refers to the overall process happening in the intestine in lieu of normal digestive breakdown of proteins, while reabsorption refers to the process occurring in the renal proximal tubule to reclaim amino and imino acids that have been filtered out of the blood via the glomerulus.
These forms of transport require energy, as the products being transported are usually moving against a higher concentration gradient. This process, called active transport, get its energy from ATP and other ATP-related cotransport systems that produce energy, like the sodium-potassium pump.
Mechanism
The primary defect associated with iminoglycinuria is a homozygous (recessive) mutation of the SLC36A2 (PAT2) gene. One of several membrane transport proteins in the solute carrier family of amino acid transporters, PAT2 is the high-affinity renal transporter of glycine, proline and hydroxyproline found to be defective in both alleles when iminoglycinuria is present in an individual. This is in contrast to the fact that when only one PAT2 allele is defective, hyperglycinuria will be present instead of iminoglycinuria. These findings delineate iminoglycinuria as the homozygous form of hyperglycinuria, with the former having a higher degree of urinary excretion of glycine and imino acids correlating to mutations in both alleles.
Another mutation suspected to convey the iminoglycinuria phenotype may be found in the SLC36A1 (PAT1) gene. Identified as the low-affinity intestinal transporter of glycine and imino acids, PAT1 works in cooperation with the renal sodium-hydrogen exchanger NHE3 (SLC9A3). As absorption and reabsorption of glycine, proline and hydroxyproline occurs through PAT1 as well, it is believed to play another role in expressing the malabsorptive iminoglycinuria phenotype. Recent reports, however, suggest a more diminished role from PAT1 in some cases of the disorder.
While PAT2 is strongly indicated as the primary mutagen responsible for iminoglycinuria, the variability of the phenotype is found to be instituted by three modifying genetic mutations. The major one among these is believed to be system IMINO.
Defined as the sodium-dependent proline transporter not inhibited by alanine, system IMINO, believed to be formed by the SLC6A20 (SIT1) gene, is a crucial mammalian transport mechanism responsible for both renal reabsorption and intestinal absorption of proline and other imino acids, such as hydroxyproline and pipecolate. The mRNA sequence for SIT1 is expressed in a great deal of the gastrointestinal tract, including the stomach, duodenum, jejunum, ileum, cecum and colon. It is also found in the kidney, optical choroid, and parts of the central nervous system such the brain and microglial cells.
Reduced penetrance is a phenomenon where a fully inherited genetic trait, such as a disease or disorder, fails to exhibit the expected phenotype. This has been reported in some cases of iminoglycinuria. Here, system IMINO is thought to play a role in reduced penetrance of iminoglycinuria by compensating for imino acid malabsorption related specifically to mutations of PAT2. Conversely, SIT1 mutations are believed to result in full expression of iminoglycinuria in some cases where heterozygous mutations of PAT2 would otherwise have only been sufficient to cause hyperglycinuria.
Two other transport systems are believed to play subsequent roles in iminoglycinuria, when mutations in them are present. The neutral amino acid transporter SLC6A19 (affecting glycine, proline, and other neutral amino acids like cysteine and tryptophan), associated with Hartnup disease, plays a role in iminoglycinuria as a modifier to PAT2 mutations and is also directly affected by the actions of SIT1. The glycine-specific transporter, SLC6A18, also has an effect on the iminoglycinuria phenotype by either compounding or compensating for failures of glycine transport.
To summarize, iminoglycinuria is primarily expressed by homozygous mutations of the PAT2 renal transporter, while the overall iminoglycinuria phenotype may be modified by normal or defective activity of SIT1 (IMINO), SLC6A19 and SLC6A18.
Diagnosis
Treament
See also
- Pipecolic acid
- Facilitated diffusion
- Oral rehydration therapy