Chronic Myelogenous Leukemia

Chronic myelogenous leukemia (CML), also known as chronic myeloid leukemia, is a cancer of the white blood cells. It is a form of leukemia characterized by the increased and unregulated growth of myeloid cells in the bone marrow and the accumulation of these cells in the blood. CML is a clonal bone marrow stem cell disorder in which a proliferation of mature granulocytes (neutrophils, eosinophils and basophils) and their precursors is found. It is a type of myeloproliferative neoplasm associated with a characteristic chromosomal translocation called the Philadelphia chromosome.
CML is largely treated with targeted drugs called tyrosine-kinase inhibitors (TKIs) which have led to dramatically improved long-term survival rates since 2001. These drugs have revolutionized treatment of this disease and allow most patients to have a good quality of life when compared to the former chemotherapy drugs. In Western countries, CML accounts for 15–25% of all adult leukemias and 14% of leukemias overall (including the pediatric population, where CML is less common).
Signs and symptoms
The way CML presents depends on the stage of the disease at diagnosis as it has been known to skip stages in some cases.
Most patients (~90%) are diagnosed during the chronic stage which is most often asymptomatic. In these cases it may be diagnosed incidentally with an elevated white blood cell count on a routine laboratory test. It can also present with symptoms indicative of hepatosplenomegaly and the resulting upper quadrant pain this causes. The enlarged spleen may put pressure on the stomach causing a loss of appetite and resulting weight loss. It may also present with mild fever and night sweats due to an elevated basal level of metabolism.
Some (<10%) are diagnosed during the accelerated stage which most often presents bleeding, petechiae and ecchymosis. In these patients fevers are most commonly the result of opportunistic infections.
Some patients are initially diagnosed in the blast phase in which the symptoms are most likely fever, bone pain and an increase in bone marrow fibrosis.
Cause
In most cases no obvious cause for CML can be isolated.
Risk factors
CML is more common in males than in females (male to female ratio of 1.4:1) and appears more commonly in the elderly with a median age at diagnosis of 65 years. Exposure to ionising radiation appears to be a risk factor, based on a 50 fold higher incidence of CML in Hiroshima and Nagasaki nuclear bombing survivors. The rate of CML in these individuals seems to peak about 10 years after the exposure.
Pathophysiology

CML was the first cancer to be linked to a clear genetic abnormality, the chromosomal translocation known as the Philadelphia chromosome. This chromosomal abnormality is so named because it was first discovered and described in 1960 by two scientists from Philadelphia, Pennsylvania, USA: Peter Nowell of the University of Pennsylvania and David Hungerford of Fox Chase Cancer Center.
In this translocation, parts of two chromosomes (the 9th and 22nd) switch places. As a result, part of the BCR ("breakpoint cluster region") gene from chromosome 22 is fused with the ABL gene on chromosome 9. This abnormal "fusion" gene generates a protein of p210 or sometimes p185 weight (p210 is short for 210 kDa protein, a shorthand used for characterizing proteins based solely on size). Because abl carries a domain that can add phosphate groups to tyrosine residues (a tyrosine kinase), the bcr-abl fusion gene product is also a tyrosine kinase.

The fused BCR-ABL protein interacts with the interleukin 3beta(c) receptor subunit. The BCR-ABL transcript is continuously active and does not require activation by other cellular messaging proteins. In turn, BCR-ABL activates a cascade of proteins that control the cell cycle, speeding up cell division. Moreover, the BCR-ABL protein inhibits DNA repair, causing genomic instability and making the cell more susceptible to developing further genetic abnormalities. The action of the BCR-ABL protein is the pathophysiologic cause of chronic myelogenous leukemia. With improved understanding of the nature of the BCR-ABL protein and its action as a tyrosine kinase, targeted therapies (the first of which was imatinib) that specifically inhibit the activity of the BCR-ABL protein have been developed. These tyrosine kinase inhibitors can induce complete remissions in CML, confirming the central importance of bcr-abl as the cause of CML.
Diagnosis

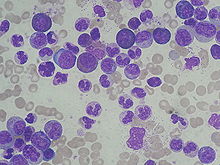

CML is often suspected on the basis of a complete blood count, which shows increased granulocytes of all types, typically including mature myeloid cells. Basophils and eosinophils are almost universally increased; this feature may help differentiate CML from a leukemoid reaction. A bone marrow biopsy is often performed as part of the evaluation for CML, and CML is diagnosed by cytogenetics that detects the translocation t(9;22)(q34;q11.2) which involves the ABL1 gene in chromosome 9 and the BCR gene in chromosome 22. As a result of this translocation, the chromosome looks smaller than its homologue chromosome, and this appearance is known as the Philadelphia chromosome chromosomal abnormality. Thus, this abnormality can be detected by routine cytogenetics, and the involved genes BCR-ABL1 can be detected by fluorescent in situ hybridization, as well as by PCR.
Controversy exists over so-called Ph-negative CML, or cases of suspected CML in which the Philadelphia chromosome cannot be detected. Many such patients in fact have complex chromosomal abnormalities that mask the (9;22) translocation, or have evidence of the translocation by FISH or RT-PCR in spite of normal routine karyotyping. The small subset of patients without detectable molecular evidence of BCR-ABL1 fusion may be better classified as having an undifferentiated myelodysplastic/myeloproliferative disorder, as their clinical course tends to be different from patients with CML.
CML must be distinguished from a leukemoid reaction, which can have a similar appearance on a blood smear.
Classification
CML is often divided into three phases based on clinical characteristics and laboratory findings. In the absence of intervention, CML typically begins in the chronic phase, and over the course of several years progresses to an accelerated phase and ultimately to a blast crisis. Blast crisis is the terminal phase of CML and clinically behaves like an acute leukemia. Drug treatment will usually stop this progression if started early. One of the drivers of the progression from chronic phase through acceleration and blast crisis is the acquisition of new chromosomal abnormalities (in addition to the Philadelphia chromosome). Some patients may already be in the accelerated phase or blast crisis by the time they are diagnosed.
Chronic phase
Approximately 85% of patients with CML are in the chronic phase at the time of diagnosis. During this phase, patients are usually asymptomatic or have only mild symptoms of fatigue, left side pain, joint and/or hip pain, or abdominal fullness. The duration of chronic phase is variable and depends on how early the disease was diagnosed as well as the therapies used. In the absence of treatment, the disease progresses to an accelerated phase. Precise patient staging based on clinical markers and personal genomic profile will likely prove beneficial in the assessment of disease history with respect to progression risk.
Accelerated phase
Criteria for diagnosing transition into the accelerated phase are somewhat variable; the most widely used criteria are those put forward by investigators at M.D. Anderson Cancer Center, by Sokal et al., and the World Health Organization. The WHO criteria are perhaps most widely used, and define the accelerated phase by the presence of ≥1 of the following haematological/cytogenetic criteria or provisional criteria concerning response to tyrosine kinase inhibitor (TKI) therapy
- Haematological/cytogenetic criteria
- Persistent or increasing high white blood cell count (> 10 × 109/L), unresponsive to therapy
- Persistent or increasing splenomegaly, unresponsive to therapy
- Persistent thrombocytosis (> 1000 × 109/L), unresponsive to therapy
- Persistent thrombocytopenia (< 100 × 109/L), unrelated to therapy
- ≥ 20% basophils in the peripheral blood
- 10―19% blasts in the peripheral blood and/or bone marrow
- Additional clonal chromosomal abnormalities in Philadelphia (Ph) chromosome-positive (Ph+) cells at diagnosis, including so-called major route abnormalities (a second Ph chromosome, trisomy 8, isochromosome 17q, trisomy 19), complex karyotype, and abnormalities of 3q26.2
- Any new clonal chromosomal abnormality in Ph+ cells that occurs during therapy
- Provisional response-to-TKI criteria
- Haematological resistance (or failure to achieve a complete haematological response d) to the first TKI
- Any haematological, cytogenetic, or molecular indications of resistance to two sequential TKIs
- Occurrence of two or more mutations in the BCR-ABL1 fusion gene during TKI therapy
The patient is considered to be in the accelerated phase if any of the above are present. The accelerated phase is significant because it signals that the disease is progressing and transformation to blast crisis is imminent. Drug treatment often becomes less effective in the advanced stages.
Blast crisis
Blast crisis is the final phase in the evolution of CML, and behaves like an acute leukemia, with rapid progression and short survival. Blast crisis is diagnosed if any of the following are present in a patient with CML:
- >20% blasts in the blood or bone marrow
- The presence of an extramedullary proliferation of blasts
Treatment
The only curative treatment for CML is a bone marrow transplant or an allogeneic stem cell transplant. Other than this there are four major mainstays of treatment in CML: treatment with tyrosine kinase inhibitors, myelosuppressive or leukopheresis therapy (to counteract the leukocytosis during early treatment), splenectomy and interferon alfa-2b treatment. Due to the high median age of patients with CML it is relatively rare for CML to be seen in pregnant women, despite this, however, chronic myelogenous leukemia can be treated with relative safety at any time during pregnancy with Interferon-alpha hormones.
Chronic phase
In the past, antimetabolites (e.g., cytarabine, hydroxyurea), alkylating agents, interferon alfa 2b, and steroids were used as treatments of CML in the chronic phase, but since the 2000s have been replaced by Bcr-Abl tyrosine-kinase inhibitors drugs that specifically target BCR-ABL, the constitutively activated tyrosine kinase fusion protein caused by the Philadelphia chromosome translocation. Despite the move to replacing cytotoxic antineoplastics (standard anticancer drugs) with tyrosine kinase inhibitors sometimes hydroxyurea is still used to counteract the high leukocyte counts encountered during treatment with tyrosine kinase inhibitors like imatinib; in these situations it may be the preferred myelosuppressive agent due to its relative lack of leukemogenic effects and hence the relative lack of potential for secondary hematologic malignancies to result from treatment. IRIS, an international study that compared interferon/cytarabine combination and the first of these new drugs imatinib, with long-term follow up, demonstrated the clear superiority of tyrosine-kinase-targeted inhibition over existing treatments.
Imatinib
The first of this new class of drugs was imatinib mesylate (marketed as Gleevec or Glivec), approved by the US Food and Drug Administration (FDA) in 2001. Imatinib was found to inhibit the progression of CML in the majority of patients (65–75%) sufficiently to achieve regrowth of their normal bone marrow stem cell population (a cytogenetic response) with stable proportions of maturing white blood cells. Because some leukemic cells (as evaluated by RT-PCR) persist in nearly all patients, the treatment has to be continued indefinitely. Since the advent of imatinib, CML has become the first cancer in which a standard medical treatment may give to the patient a normal life expectancy.
Dasatinib, nilotinib, radotinib and bosutinib
To overcome imatinib resistance and to increase responsiveness to TK inhibitors, four novel agents were later developed. The first, dasatinib, blocks several further oncogenic proteins, in addition to more potent inhibition of the BCR-ABL protein, and was initially approved in 2007 by the US FDA to treat CML in patients who were either resistant to or intolerant of imatinib. A second new TK inhibitor, nilotinib, was also approved by the FDA for the same indication. In 2010, nilotinib and dasatinib were also approved for first-line therapy, making three drugs in this class available for treatment of newly diagnosed CML. In 2012, Radotinib joined the class of novel agents in the inhibition of the BCR-ABL protein and was approved in South Korea for patients resistant to or intolerant of imatinib. Bosutinib received US FDA and EU European Medicines Agency approval on 4 September 2012 and 27 March 2013 respectively for the treatment of adult patients with Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML) with resistance, or intolerance to prior therapy.
Treatment-resistant CML
While capable of producing significantly improved responses compared with the action of imatinib, neither dasatinib nor nilotinib could overcome drug resistance caused by one particular mutation found to occur in the structure of BCR-ABL1 known as the T315I mutation (in other words, where the 315th amino acid is mutated from a threonine residue to an isoleucine residue). Two approaches were developed to the treatment of CML as a result:
In 2007, Chemgenex released results of an open-label Phase 2/3 study (CGX-635-CML-202) that investigated the use of a non BCR-ABL targeted agent omacetaxine, administered subcutaneously (under the skin) in patients who had failed with imatinib and exhibited T315I kinase domain mutation. This is a study which is ongoing through 2014. In September 2012, the FDA approved omacetaxine for the treatment of CML in the case of resistance to other chemotherapeutic agents.
Independently, ARIAD pharmaceuticals, adapting the chemical structures from first and second-generation TK inhibitors, arrived at a new pan-BCR-ABL1 inhibitor which showed (for the first time) efficacy against T315I, as well as all other known mutations of the oncoprotein. The drug, ponatinib, gained FDA approval in December 2012 for treatment of patients with resistant or intolerant CML. Just as with second generation TK inhibitors, early approval is being sought to extend the use of ponatinib to newly diagnosed CML also.
Vaccination
In 2005, encouraging but mixed results of vaccination were reported with the BCR/ABL1 p210 fusion protein in patients with stable disease, with GM-CSF as an adjuvant.
Prognosis
Before the advent of tyrosine kinase inhibitors, the median survival time for CML patients had been about 3–5 years from time of diagnosis.
With the use of tyrosine kinase inhibitors, survival rates have improved dramatically. A 2006 followup of 553 patients using imatinib (Gleevec) found an overall survival rate of 89% after five years.
A 2011 followup of 832 patients using imatinib who achieved a stable cytogenetic response found an overall survival rate of 95.2% after 8 years, which is similar to the rate in the general population. Fewer than 1% of patients died because of leukemia progression.
Epidemiology
United Kingdom
CML accounts for 8% of all leukaemias in the UK, and around 680 people were diagnosed with the disease in 2011.
United States
The American Cancer Society estimates that in 2014, about 5,980 new cases of chronic myeloid leukemia were diagnosed, and about 810 people died of the disease. This means that a little over 10% of all newly diagnosed leukemia cases will be chronic myeloid leukemia. The average risk of a person getting this disease is 1 in 588. The disease is more common in men than women, and more common in whites than African-Americans. The average age at diagnosis is 64 years, and this disease is rarely seen in children.