Malignant Rhabdoid Tumour
Malignant rhabdoid tumour (MRT) is a very aggressive form of tumour originally described as a variant of Wilms' tumour, which is primarily a kidney tumour that occurs mainly in children.
MRT was first described as a variant of Wilms' tumour of the kidney in 1978. MRTs are a rare and highly malignant childhood neoplasm. Rhabdoid tumours outside the kidney were later reported in many tissues including the liver, soft tissue, and the central nervous system. Several cases of primary intracranial MRT have been reported since its recognition as a separate entity in 1978. The term rhabdoid was used due to its similarity with rhabdomyosarcoma under the light microscope. The exact pathogenesis of MRT is unknown.
The cerebellum is the most common location for primary intracerebral MRT (i.e., atypical teratoid rhabdoid tumor). Biggs et al. were first to report a primary intracranial MRT around 1987.
Although the cell of origin is not known, cytogenetic studies have suggested a common genetic basis for rhabdoid tumours regardless of location with abnormalities in chromosome 22 commonly occurring.
Genetics
There have been reported cases of a child having both atypical teratoid rhabdoid tumours in the brain as well as rhabdoid tumours of the kidney. Weeks and associates reported on 111 renal rhabdoid cases of which 13.5% also had a central nervous system malignancy. It has been hypothesized that a germline INI mutation may predispose a child to these tumours. There have been some references in the literature alluding to a new diagnosis called rhabdoid predisposition syndrome related to the gene hSNF5/INI1.
These tumours have been associated with mutations in the SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1) located on the long arm of chromosome 22 (22q11) and transcription activator BRG1/ATP-dependent chromatin remodeler (SMARCA4) located on the short arm of chromosome 19 (19p13.2).
Rhabdoid tumours in kidney and brain
Considerable debate has been focused on whether atypical teratoid rhabdoid tumors are the same as rhabdoid tumours of the kidney (i.e., just extrarenal MRTs). The recent recognition that both CNS atypical teratoid/rhabdoid tumours and MRTs have deletions of the INI1 gene in chromosome 22 indicates that rhabdoid tumours of the kidney and brain are identical or closely related entities, although the CNS variant tends to have its mutations on Taxon 9 and MRTs elsewhere. This observation is not surprising because rhabdoid tumours at both locations possess similar histologic, clinical, and demographic features. Moreover, 10-15% of patients with MRTs have synchronous or metachronous brain tumours, many of which are second primary malignant rhabdoid tumours. This similarity excludes composite rhabdoid tumours, which occur mainly in adults.
Diagnosis
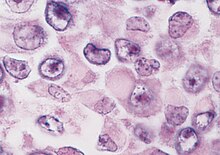
The histologic diagnosis of malignant rhabdoid tumour depends on identification of characteristic rhabdoid cells—large cells with eccentrically located nuclei and abundant, eosinophilic cytoplasm. However, the histology can be heterogeneous and the diagnosis of MRT can often be difficult. Misclassifications can occur.
In MRTs, the INI1 gene (SMARCB1) on chromosome 22q functions as a classic tumour suppressor gene. Inactivation of INI1 can occur via deletion, mutation, or acquired UPD.
In a recent study, Single nucleotide polymorphism array karyotyping identified deletions or LOH of 22q in 49/51 rhabdoid tumours. Of these, 14 were copy neutral LOH (or acquired UPD), which is detectable by SNP array karyotyping, but not by FISH, cytogenetics, or array CGH. MLPA detected a single exon homozygous deletion in one sample that was below the resolution of the SNP array. SNP array karyotyping can be used to distinguish, for example, a medulloblastoma with an isochromosome 17q from a primary rhabdoid tumour with loss of 22q11.2. When indicated, molecular analysis of INI1 using MLPA and direct sequencing may then be employed. Once the tumour-associated changes are found, an analysis of germline DNA from the patient and the parents can be done to rule out an inherited or de novo germline mutation or deletion of INI1, so that appropriate recurrence risk assessments can be made.
Prognosis
Regardless of location, all rhabdoid tumours are highly aggressive, have a poor prognosis, and tend to occur in children less than two years of age.
See also
- Epithelioid sarcoma
- Atypical teratoid rhabdoid tumour