Acute Myeloid Leukemia

Acute myeloid leukemia (AML) is a cancer of the myeloid line of blood cells, characterized by the rapid growth of abnormal cells that build up in the bone marrow and blood and interfere with normal blood cell production. Symptoms may include feeling tired, shortness of breath, easy bruising and bleeding, and increased risk of infection. Occasionally, spread may occur to the brain, skin, or gums. As an acute leukemia, AML progresses rapidly and is typically fatal within weeks or months if left untreated.
Risk factors include smoking, previous chemotherapy or radiation therapy, myelodysplastic syndrome, and exposure to the chemical benzene. The underlying mechanism involves replacement of normal bone marrow with leukemia cells, which results in a drop in red blood cells, platelets, and normal white blood cells. Diagnosis is generally based on bone marrow aspiration and specific blood tests. AML has several subtypes for which treatments and outcomes may vary.
AML typically is initially treated with chemotherapy, with the aim of inducing remission. People may then go on to receive additional chemotherapy, radiation therapy, or a stem cell transplant. The specific genetic mutations present within the cancer cells may guide therapy, as well as determine how long that person is likely to survive.
In 2015, AML affected about one million people and resulted in 147,000 deaths globally. It most commonly occurs in older adults. Males are affected more often than females. The five-year survival rate is about 35% in people under 60 years old and 10% in people over 60 years old. Older people whose health is too poor for intensive chemotherapy have a typical survival of five to ten months. It accounts for roughly 1.8% of cancer deaths in the United States.
Signs and symptoms

Most signs and symptoms of AML are caused by the replacement of normal blood cells with leukemic cells. A lack of normal white blood cell production makes people more susceptible to infections; while the leukemic cells themselves are derived from white blood cell precursors, they have no infection-fighting capacity. A drop in red blood cell count (anemia) can cause fatigue, paleness, and shortness of breath. A lack of platelets can lead to easy bruising or bleeding with minor trauma.
The early signs of AML are often vague and nonspecific, and may be similar to those of influenza or other common illnesses. Some generalized symptoms include fever, fatigue, weight loss or loss of appetite, shortness of breath, anemia, easy bruising or bleeding, petechiae (flat, pin-head sized spots under the skin caused by bleeding), bone and joint pain, and persistent or frequent infections.
Enlargement of the spleen may occur in AML, but it is typically mild and asymptomatic. Lymph node swelling is rare in AML, in contrast to acute lymphoblastic leukemia. The skin is involved about 10% of the time in the form of leukemia cutis. Rarely, Sweet's syndrome, a paraneoplastic inflammation of the skin, can occur with AML.
Some people with AML may experience swelling of the gums because of infiltration of leukemic cells into the gum tissue. Rarely, the first sign of leukemia may be the development of a solid leukemic mass or tumor outside of the bone marrow, called a chloroma. Occasionally, a person may show no symptoms, and the leukemia may be discovered incidentally during a routine blood test.
Risk factors
A number of risk factors for developing AML have been identified, including: other blood disorders, chemical exposures, ionizing radiation, and genetics.
Other blood disorders
"Preleukemic" blood disorders, such as myelodysplastic syndrome (MDS) or myeloproliferative neoplasms (MPN), can evolve into AML; the exact risk depends on the type of MDS/MPN. The presence of asymptomatic clonal hematopoiesis also raises the risk of transformation into AML to 0.5–1.0% per year.
Chemical exposure
Exposure to anticancer chemotherapy, in particular alkylating agents, can increase the risk of subsequently developing AML. The risk is highest about three to five years after chemotherapy. Other chemotherapy agents, specifically epipodophyllotoxins and anthracyclines, have also been associated with treatment-related leukemias, which are often associated with specific chromosomal abnormalities in the leukemic cells.
Occupational chemical exposure to benzene and other aromatic organic solvents is controversial as a cause of AML. Benzene and many of its derivatives are known to be carcinogenic in vitro. While some studies have suggested a link between occupational exposure to benzene and increased risk of AML, others have suggested the attributable risk, if any, is slight.
Radiation
High amounts of ionizing radiation exposure can increase the risk of AML. Survivors of the atomic bombings of Hiroshima and Nagasaki had an increased rate of AML, as did radiologists exposed to high levels of X-rays prior to the adoption of modern radiation safety practices. People treated with ionizing radiation after treatment for prostate cancer, non-Hodgkin lymphoma, lung cancer, and breast cancer have the highest chance of acquiring AML, but this increased risk returns to the background risk observed in the general population after 12 years.
Genetics
A hereditary risk for AML appears to exist. Multiple cases of AML developing in a family at a rate higher than predicted by chance alone have been reported. Several congenital conditions may increase the risk of leukemia; the most common is probably Down syndrome, which is associated with a 10- to 18-fold increase in the risk of AML. In a second example, inactivating mutations in one of the two parental GATA2 genes lead to a reduction, i.e. a haploinsufficiency, in the cellular levels of the gene's product, the GATA2 transcription factor, and thereby to a rare autosomal dominant genetic disease, GATA2 deficiency. This disease is associated with a highly variable set of disorders including an exceedingly high risk of developing AML. The specific genetic abnormalities causing AML usually vary between those who develop the disease as a child versus an adult. However, GATA2 deficiency-induced AML may first appear in children or adults.
Diagnosis

The first clue to a diagnosis of AML is typically an abnormal result on a complete blood count. While an excess of abnormal white blood cells (leukocytosis) is a common finding with the leukemia, and leukemic blasts are sometimes seen, AML can also present with isolated decreases in platelets, red blood cells, or even with a low white blood cell count (leukopenia). While a presumptive diagnosis of AML can be made by examination of the peripheral blood smear when there are circulating leukemic blasts, a definitive diagnosis usually requires an adequate bone marrow aspiration and biopsy as well as ruling out pernicious anemia (Vitamin B12 deficiency), folic acid deficiency and copper deficiency.
Marrow or blood is examined under light microscopy, as well as flow cytometry, to diagnose the presence of leukemia, to differentiate AML from other types of leukemia (e.g. acute lymphoblastic leukemia – ALL), and to classify the subtype of disease. A sample of marrow or blood is typically also tested for chromosomal abnormalities by routine cytogenetics or fluorescent in situ hybridization. Genetic studies may also be performed to look for specific mutations in genes such as FLT3, nucleophosmin, and KIT, which may influence the outcome of the disease.
Cytochemical stains on blood and bone marrow smears are helpful in the distinction of AML from ALL, and in subclassification of AML. The combination of a myeloperoxidase or Sudan black stain and a nonspecific esterase stain will provide the desired information in most cases. The myeloperoxidase or Sudan black reactions are most useful in establishing the identity of AML and distinguishing it from ALL. The nonspecific esterase stain is used to identify a monocytic component in AMLs and to distinguish a poorly differentiated monoblastic leukemia from ALL.
The diagnosis and classification of AML can be challenging, and should be performed by a qualified hematopathologist or hematologist. In straightforward cases, the presence of certain morphologic features (such as Auer rods) or specific flow cytometry results can distinguish AML from other leukemias; however, in the absence of such features, diagnosis may be more difficult.
The two most commonly used classification schemata for AML are the older French-American-British (FAB) system and the newer World Health Organization (WHO) system. According to the widely used WHO criteria, the diagnosis of AML is established by demonstrating involvement of more than 20% of the blood and/or bone marrow by leukemic myeloblasts, except in the three best prognosis forms of acute myeloid leukemia with recurrent genetic abnormalities (t(8;21), inv(16), and t(15;17)) in which the presence of the genetic abnormality is diagnostic irrespective of blast percent. The FAB classification is a bit more stringent, requiring a blast percentage of at least 30% in bone marrow or peripheral blood for the diagnosis of AML. AML must be carefully differentiated from "preleukemic" conditions such as myelodysplastic or myeloproliferative syndromes, which are treated differently.
Because acute promyelocytic leukemia (APL) has the highest curability and requires a unique form of treatment, it is important to quickly establish or exclude the diagnosis of this subtype of leukemia. Fluorescent in situ hybridization performed on blood or bone marrow is often used for this purpose, as it readily identifies the chromosomal translocation [t(15;17)(q22;q12);] that characterizes APL. There is also a need to molecularly detect the presence of PML/RARA fusion protein, which is an oncogenic product of that translocation.
World Health Organization
The WHO 2008 classification of AML attempts to be more clinically useful and to produce more meaningful prognostic information than the FAB criteria. Each of the WHO categories contains numerous descriptive subcategories of interest to the hematopathologist and oncologist; however, most of the clinically significant information in the WHO schema is communicated via categorization into one of the subtypes listed below.
The WHO subtypes of AML are:
| Name | Description | ICD-O |
|---|---|---|
| Acute myeloid leukemia with recurrent genetic abnormalities | Includes:
|
Multiple |
| AML with myelodysplasia-related changes | This category includes people who have had a prior documented myelodysplastic syndrome (MDS) or myeloproliferative disease (MPD) that then has transformed into AML, or who have cytogenetic abnormalities characteristic for this type of AML (with previous history of MDS or MPD that has gone unnoticed in the past, but the cytogenetics is still suggestive of MDS/MPD history). This category of AML occurs most often in elderly people and often has a worse prognosis. Includes:
|
M9895/3 |
| Therapy-related myeloid neoplasms | This category includes people who have had prior chemotherapy and/or radiation and subsequently develop AML or MDS. These leukemias may be characterized by specific chromosomal abnormalities, and often carry a worse prognosis. | M9920/3 |
| Myeloid sarcoma | This category includes myeloid sarcoma. | |
| Myeloid proliferations related to Down syndrome | This category includes so-called "transient abnormal myelopoiesis" and "myeloid leukemia associated with Down syndrome" | |
| Blastic plasmacytoid dendritic cell neoplasm | This category includes so-called "blastic plasmacytoid dendritic cell neoplasm" | |
| AML not otherwise categorized | Includes subtypes of AML that do not fall into the above categories
|
M9861/3 |
Acute leukemias of ambiguous lineage (also known as mixed phenotype or biphenotypic acute leukemia) occur when the leukemic cells can not be classified as either myeloid or lymphoid cells, or where both types of cells are present.
French-American-British
The French-American-British (FAB) classification system divides AML into eight subtypes, M0 through to M7, based on the type of cell from which the leukemia developed and its degree of maturity. AML of types M0 to M2 may be called acute myeloblastic leukemia. Classification is done by examining the appearance of the malignant cells with light microscopy and/or by using cytogenetics to characterize any underlying chromosomal abnormalities. The subtypes have varying prognoses and responses to therapy. Although the WHO classification (see above) may be more useful, the FAB system is still widely used.
Six FAB subtypes (M1 through to M6) were initially proposed in 1976, although later revisions added M7 in 1985 and M0 in 1987.
| Type | Name | Cytogenetics | Percentage of adults with AML | Immunophenotype | ||||
|---|---|---|---|---|---|---|---|---|
| CD14 | CD15 | CD33 | HLA-DR | Other | ||||
| M0 | acute myeloblastic leukemia, minimally differentiated | 5% | − | − | + | + | MPO − | |
| M1 | acute myeloblastic leukemia, without maturation | 15% | − | − | + | + | MPO + | |
| M2 | acute myeloblastic leukemia, with granulocytic maturation | t(8;21)(q22;q22), t(6;9) | 25% | − | + | + | + | |
| M3 | promyelocytic, or acute promyelocytic leukemia (APL) | t(15;17) | 10% | − | + | + | − | |
| M4 | acute myelomonocytic leukemia | inv(16)(p13q22), del(16q) | 20% | <45% | + | + | + | |
| M4eo | myelomonocytic together with bone marrow eosinophilia | inv(16), t(16;16) | 5% | +/− | + | + | CD2+ | |
| M5 | acute monoblastic leukemia (M5a) or acute monocytic leukemia (M5b) | del (11q), t(9;11), t(11;19) | 10% | >55% | + | + | + | |
| M6 | acute erythroid leukemias, including erythroleukemia (M6a) and very rare pure erythroid leukemia (M6b) | 5% | − | +/− | +/− | +/− | Glycophorin + | |
| M7 | acute megakaryoblastic leukemia | t(1;22) | 5% | − | − | + | +/− | CD41/CD61+ |
The morphologic subtypes of AML also include rare types not included in the FAB system, such as acute basophilic leukemia, which was proposed as a ninth subtype, M8, in 1999.
Pathophysiology

The malignant cell in AML is the myeloblast. In normal hematopoiesis, the myeloblast is an immature precursor of myeloid white blood cells; a normal myeloblast will gradually mature into a mature white blood cell. In AML, though, a single myeloblast accumulates genetic changes which "freeze" the cell in its immature state and prevent differentiation. Such a mutation alone does not cause leukemia; however, when such a "differentiation arrest" is combined with other mutations which disrupt genes controlling proliferation, the result is the uncontrolled growth of an immature clone of cells, leading to the clinical entity of AML.
Much of the diversity and heterogeneity of AML is because leukemic transformation can occur at a number of different steps along the differentiation pathway. Modern classification schemes for AML recognize that the characteristics and behavior of the leukemic cell (and the leukemia) may depend on the stage at which differentiation was halted.
Specific cytogenetic abnormalities can be found in many people with AML; the types of chromosomal abnormalities often have prognostic significance. The chromosomal translocations encode abnormal fusion proteins, usually transcription factors whose altered properties may cause the "differentiation arrest". For example, in APL, the t(15;17) translocation produces a PML-RARA fusion protein which binds to the retinoic acid receptor element in the promoters of several myeloid-specific genes and inhibits myeloid differentiation.
The clinical signs and symptoms of AML result from the growth of leukemic clone cells, which tends to interfere with the development of normal blood cells in the bone marrow. This leads to neutropenia, anemia, and thrombocytopenia. The symptoms of AML are, in turn, often due to the low numbers of these normal blood elements. In rare cases, people with AML can develop a chloroma, or solid tumor of leukemic cells outside the bone marrow, which can cause various symptoms depending on its location.
An important pathophysiological mechanism of leukemogenesis in AML is the epigenetic induction of dedifferentiation by genetic mutations that alter the function of epigenetic enzymes, such as the DNA demethylase TET2 and the metabolic enzymes IDH1 and IDH2, which lead to the generation of a novel oncometabolite, D-2-hydroxyglutarate, which inhibits the activity of epigenetic enzymes such as TET2. The hypothesis is that such epigenetic mutations lead to the silencing of tumor suppressor genes and/or the activation of proto-oncogenes.
Treatment
First-line treatment of AML consists primarily of chemotherapy, and is divided into two phases: induction and postremission (or consolidation) therapy. The goal of induction therapy is to achieve a complete remission by reducing the number of leukemic cells to an undetectable level; the goal of consolidation therapy is to eliminate any residual undetectable disease and achieve a cure. Hematopoietic stem cell transplantation is usually considered if induction chemotherapy fails or after a person relapses, although transplantation is also sometimes used as front-line therapy for people with high-risk disease. Efforts to use tyrosine kinase inhibitors in AML continue.
Induction
All FAB subtypes except M3 are usually given induction chemotherapy with cytarabine (ara-C) and an anthracycline (most often daunorubicin). This induction chemotherapy regimen is known as "7+3" (or "3+7"), because the cytarabine is given as a continuous IV infusion for seven consecutive days while the anthracycline is given for three consecutive days as an IV push. Up to 70% of people with AML will achieve a remission with this protocol. Other alternative induction regimens, including high-dose cytarabine alone, FLAG-like regimens or investigational agents, may also be used. Because of the toxic effects of therapy, including myelosuppression and an increased risk of infection, induction chemotherapy may not be offered to the very elderly, and the options may include less intense chemotherapy or palliative care.
The M3 subtype of AML, also known as acute promyelocytic leukemia (APL), is treated with either arsenic trioxide (ATO) monotherapy, or the drug all-trans-retinoic acid (ATRA) in addition to induction chemotherapy, usually an anthracycline. Care must be taken to prevent disseminated intravascular coagulation (DIC), complicating the treatment of APL when the promyelocytes release the contents of their granules into the peripheral circulation. APL is eminently curable, with well-documented treatment protocols.
The goal of the induction phase is to reach a complete remission. Complete remission does not mean the disease has been cured; rather, it signifies no disease can be detected with available diagnostic methods. Complete remission is obtained in about 50%–75% of newly diagnosed adults, although this may vary based on the prognostic factors described above. The length of remission depends on the prognostic features of the original leukemia. In general, all remissions will fail without additional consolidation therapy.
There is insufficient evidence to determine if prescribing all-trans retinoic acid (ATRA) in addition to chemotherapy to adults that suffer from acute myeloid leukaemia is helpful.
Consolidation
Even after complete remission is achieved, leukemic cells likely remain in numbers too small to be detected with current diagnostic techniques. If no further postremission or consolidation therapy is given, almost all people with AML will eventually relapse. Therefore, more therapy is necessary to eliminate nondetectable disease and prevent relapse – that is, to achieve a cure.
The specific type of postremission therapy is individualized based on a person's prognostic factors (see above) and general health. For good-prognosis leukemias (i.e. inv(16), t(8;21), and t(15;17)), people will typically undergo an additional three to five courses of intensive chemotherapy, known as consolidation chemotherapy. For people at high risk of relapse (e.g. those with high-risk cytogenetics, underlying MDS, or therapy-related AML), allogeneic stem cell transplantation is usually recommended if the person is able to tolerate a transplant and has a suitable donor. The best postremission therapy for intermediate-risk AML (normal cytogenetics or cytogenetic changes not falling into good-risk or high-risk groups) is less clear and depends on the specific situation, including the age and overall health of the person, the person's values, and whether a suitable stem cell donor is available.
For people who are not eligible for a stem cell transplant, immunotherapy with a combination of histamine dihydrochloride (Ceplene) and interleukin 2 (Proleukin) after the completion of consolidation has been shown to reduce the absolute relapse risk by 14%, translating to a 50% increase in the likelihood of maintained remission.
Relapsed AML
For people with relapsed AML, the only proven potentially curative therapy is a hematopoietic stem cell transplant, if one has not already been performed. In 2000, the monoclonal antibody-linked cytotoxic agent gemtuzumab ozogamicin (Mylotarg) was approved in the United States for people aged more than 60 years with relapsed AML who are not candidates for high-dose chemotherapy. This drug was voluntarily withdrawn from the market by its manufacturer, Pfizer in 2010, but newer data aided its reintroduction in 2017.
Since treatment options for relapsed AML are so limited, palliative care or enrollment in a clinical trial may be offered.
Supportive treatment
Adding aerobic physical exercises to the standard of care may result in little to no difference in the mortality, in the quality of life and in the physical functioning. These exercises may result in a slight reduction in depression. Furthermore, aerobic physical exercises probably reduce fatigue.
Side effects
Treatments for AML like chemotherapy or stem cell transplant can trigger side effects. People that receive a stem cell transplant are at risk for developing a graft-versus-host disease, and suffer from bleeding events that may require platelet transfusions.
Prognosis
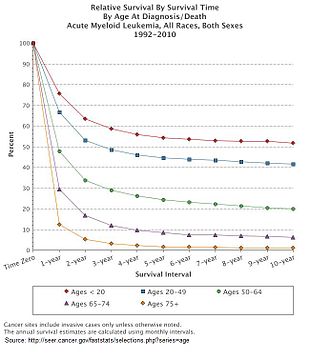

AML is a curable disease. The chance of cure for a specific person depends on a number of prognostic factors.
Cytogenetics
The single most important prognostic factor in AML is cytogenetics, or the chromosomal structure of the leukemic cell. Certain cytogenetic abnormalities are associated with very good outcomes (for example, the (15;17) translocation in APL). About half of people with AML have "normal" cytogenetics; they fall into an intermediate risk group. A number of other cytogenetic abnormalities are known to associate with a poor prognosis and a high risk of relapse after treatment.
The first publication to address cytogenetics and prognosis was the MRC trial of 1998:
| Risk Category | Abnormality | Five-year survival | Relapse rate |
|---|---|---|---|
| Good | t(8;21), t(15;17), inv(16) | 70% | 33% |
| Intermediate | Normal, +8, +21, +22, del(7q), del(9q), Abnormal 11q23, all other structural or numerical changes | 48% | 50% |
| Poor | −5, −7, del(5q), Abnormal 3q, Complex cytogenetics | 15% | 78% |
Later, the Southwest Oncology Group and Eastern Cooperative Oncology Group and, later still, Cancer and Leukemia Group B published other, mostly overlapping lists of cytogenetics prognostication in leukemia.
Myelodysplastic syndrome
AML arising from a pre-existing myelodysplastic syndrome (MDS) or myeloproliferative disease (so-called secondary AML) has a worse prognosis, as does treatment-related AML arising after chemotherapy for another previous malignancy. Both of these entities are associated with a high rate of unfavorable cytogenetic abnormalities.
Other prognostic markers
In some studies, age >60 years and elevated lactate dehydrogenase level were also associated with poorer outcomes. As with most forms of cancer, performance status (i.e. the general physical condition and activity level of the person) plays a major role in prognosis as well.
The five-year survival rate is about 25% overall. Age plays a significant role: 40% of people under the age of 60, but just 10% of those over it, live five years after diagnosis.
Genotype
A large number of molecular alterations are under study for their prognostic impact in AML. However, only FLT3-ITD, NPM1, CEBPA and c-KIT are currently included in validated international risk stratification schema. These are expected to increase rapidly in the near future. FLT3 internal tandem duplications (ITDs) have been shown to confer a poorer prognosis in AML with normal cytogenetics. Several FLT3 inhibitors have undergone clinical trials, with mixed results. Two other mutations – NPM1 and biallelic CEBPA are associated with improved outcomes, especially in people with normal cytogenetics and are used in current risk stratification algorithms.
Researchers are investigating the clinical significance of c-KIT mutations in AML. These are prevalent, and potentially clinically relevant because of the availability of tyrosine kinase inhibitors, such as imatinib and sunitinib that can block the activity of c-KIT pharmacologically. It is expected that additional markers (e.g., RUNX1, ASXL1, and TP53) that have consistently been associated with an inferior outcome will soon be included in these recommendations. The prognostic importance of other mutated genes (e.g., DNMT3A, IDH1, IDH2) is less clear.
Expectation of cure
Cure rates in clinical trials have ranged from 20 to 45%; although clinical trials often include only younger people and those able to tolerate aggressive therapies. The overall cure rate for all people with AML (including the elderly and those unable to tolerate aggressive therapy) is likely lower. Cure rates for APL can be as high as 98%.
Relapse
Relapse is common, and the prognosis is poor. Long-term survival after a relapse is so rare that the only known case was submitted to the Catholic Church as evidence of a miracle attributed to Marie-Marguerite d'Youville.
Epidemiology
AML is a relatively rare cancer. There are approximately 10,500 new cases each year in the United States, and the incidence rate has remained stable from 1995 through 2005. AML accounts for 1.2% of all cancer deaths in the United States.
The incidence of AML increases with age; the median age at diagnosis is 63 years. AML accounts for about 90% of all acute leukemias in adults, but is rare in children. The rate of therapy-related AML (that is, AML caused by previous chemotherapy) is rising; therapy-related disease currently accounts for about 10–20% of all cases of AML. AML is slightly more common in men, with a male-to-female ratio of 1.3:1.
There is some geographic variation in the incidence of AML. In adults, the highest rates are seen in North America, Europe, and Oceania, while adult AML is rarer in Asia and Latin America. In contrast, childhood AML is less common in North America and India than in other parts of Asia. These differences may be due to population genetics, environmental factors, or a combination of the two.
AML accounts for 34% of all leukemia cases in the UK, and around 2,900 people were diagnosed with the disease in 2011.
History

The first published description of a case of leukemia in medical literature dates to 1827 when French physician Alfred-Armand-Louis-Marie Velpeau described a 63-year-old florist who developed an illness characterized by fever, weakness, urinary stones, and substantial enlargement of the liver and spleen. Velpeau noted the blood of this person had a consistency "like gruel", and speculated the appearance of the blood was due to white corpuscles.:1071 In 1845, a series of people who died with enlarged spleens and changes in the "colors and consistencies of their blood" was reported by the Edinburgh-based pathologist J.H. Bennett; he used the term "leucocythemia" to describe this pathological condition.
The term "leukemia" was coined by Rudolf Virchow, the renowned German pathologist, in 1856. As a pioneer in the use of the light microscope in pathology, Virchow was the first to describe the abnormal excess of white blood cells in people with the clinical syndrome described by Velpeau and Bennett. As Virchow was uncertain of the etiology of the white blood cell excess, he used the purely descriptive term "leukemia" (Greek: "white blood") to refer to the condition.
Further advances in the understanding of AML occurred rapidly with the development of new technology. In 1877, Paul Ehrlich developed a technique of staining blood films which allowed him to describe in detail normal and abnormal white blood cells. Wilhelm Ebstein introduced the term "acute leukemia" in 1889 to differentiate rapidly progressive and fatal leukemias from the more indolent chronic leukemias. The term "myeloid" was coined by Franz Ernst Christian Neumann in 1869, as he was the first to recognize white blood cells were made in the bone marrow (Greek: μυєλός, myelos, lit. '(bone) marrow') as opposed to the spleen. The technique of bone marrow examination to diagnose leukemia was first described in 1879 by Mosler. Finally, in 1900, the myeloblast, which is the malignant cell in AML, was characterized by Otto Naegeli, who divided the leukemias into myeloid and lymphocytic.
In 2008, AML became the first cancer genome to be fully sequenced. DNA extracted from leukemic cells were compared to unaffected skin. The leukemic cells contained acquired mutations in several genes that had not previously been associated with the disease.
Pregnancy
Leukemia is rarely associated with pregnancy, affecting only about 1 in 10,000 pregnant women. How it is handled depends primarily on the type of leukemia. Acute leukemias normally require prompt, aggressive treatment, despite significant risks of pregnancy loss and birth defects, especially if chemotherapy is given during the developmentally sensitive first trimester.