Pulmonary Hypertension

Pulmonary hypertension (PH or PHTN) is a condition of increased blood pressure within the arteries of the lungs. Symptoms include shortness of breath, syncope, tiredness, chest pain, swelling of the legs, and a fast heartbeat. The condition may make it difficult to exercise. Onset is typically gradual.
A patient is deemed to have pulmonary hypertension if the pulmonary mean arterial pressure is greater than 25mmHg at rest, or greater than 30mmHg during exercise.
The cause is often unknown. Risk factors include a family history, prior blood clots in the lungs, HIV/AIDS, sickle cell disease, cocaine use, chronic obstructive pulmonary disease, sleep apnea, living at high altitudes, and problems with the mitral valve. The underlying mechanism typically involves inflammation and subsequent remodeling of the arteries in the lungs. Diagnosis involves first ruling out other potential causes.
There is currently no cure for pulmonary hypertension, although research on a cure is ongoing. Treatment depends on the type of disease. A number of supportive measures such as oxygen therapy, diuretics, and medications to inhibit blood clotting may be used. Medications specifically used to treat pulmonary hypertension include epoprostenol, treprostinil, iloprost, bosentan, ambrisentan, macitentan, and sildenafil. Lung transplantation may be an option in severe cases.
While the exact frequency of the condition is unknown, it is estimated that about 1,000 new cases occur a year in the United States. Females are more often affected than males. Onset is typically between 20 and 60 years of age. It was first identified by Ernst von Romberg in 1891.
Signs and symptoms
The symptoms of pulmonary hypertension include the following:
- Shortness of breath
- Fatigue
- Chest pain
- Palpitations (heartbeat rate increased)
- Right-sided abdominal pain
- Poor appetite
- Lightheadedness
- Fainting
- Swelling (legs/ankles)
- Cyanosis
Less common signs/symptoms include non-productive cough and exercise-induced nausea and vomiting. Coughing up of blood may occur in some patients, particularly those with specific subtypes of pulmonary hypertension such as heritable pulmonary arterial hypertension, Eisenmenger syndrome and chronic thromboembolic pulmonary hypertension. Pulmonary venous hypertension typically presents with shortness of breath while lying flat or sleeping (orthopnea or paroxysmal nocturnal dyspnea), while pulmonary arterial hypertension (PAH) typically does not.
Other typical signs of pulmonary hypertension include an accentuated pulmonary component of the second heart sound, a right ventricular third heart sound, and parasternal heave indicating a hypertrophied right ventricle. Signs of systemic congestion resulting from right-sided heart failure include jugular venous distension, ascites, and hepatojugular reflux. Evidence of tricuspid insufficiency and pulmonic regurgitation is also sought and, if present, is consistent with the presence of pulmonary hypertension.
Causes
Pulmonary hypertension is a pathophysiologic condition with many possible causes. Indeed, this condition frequently accompanies severe heart or lung conditions. A 1973 World Health Organization meeting was the first attempt to classify pulmonary hypertension by its cause, and a distinction was made between primary PH (resulting from a disease of the pulmonary arteries) and secondary PH (resulting secondary to other, non-vascular causes). Further, primary PH was divided into the "arterial plexiform", "veno-occlusive" and "thromboembolic" forms. In 1998, a second conference at Évian-les-Bains addressed the causes of secondary PH. Subsequent third, fourth, and fifth (2013) World Symposia on PAH have further defined the classification of PH. The classification continues to evolve based on improved understanding of the disease mechanisms.
Most recently in 2015, the WHO guidelines were updated by the European Society of Cardiology (ESC) and European Respiratory Society (ERS). These guidelines are endorsed by the International Society for Heart and Lung Transplantation, and provide the current framework for understanding and treatment of pulmonary hypertension.
Classification
According to WHO classification there are 5 groups of PH, where Group I (pulmonary arterial hypertension) is further subdivided into Group I' and Group I'' classes. The most recent WHO classification system (with adaptations from the more recent ESC/ERS guidelines shown in italics) can be summarized as follows:
WHO Group I – Pulmonary arterial hypertension (PAH)
- Idiopathic
- Heritable (BMPR2, ALK1, SMAD9, caveolin 1, KCNK3 mutations)
- Drug- and toxin-induced (e.g., methamphetamine use)
- Associated conditions:Connective tissue disease, HIV infection, Portal hypertension, Congenital heart diseases, Schistosomiasis
WHO Group I' – Pulmonary veno-occlusive disease (PVOD), pulmonary capillary hemangiomatosis (PCH)
- Idiopathic
- Heritable (EIF2AK4 mutations)
- Drugs, toxins and radiation-induced
- Associated conditions:connective tissue disease, HIV infection
WHO Group I" – Persistent pulmonary hypertension of the newborn
WHO Group II – Pulmonary hypertension secondary to left heart disease
- Left ventricular systolic dysfunction
- Left ventricular diastolic dysfunction
- Valvular heart disease
- Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathy
- Congenital/acquired pulmonary venous stenosis
WHO Group III – Pulmonary hypertension due to lung disease, chronic hypoxia
- Chronic obstructive pulmonary disease (COPD)
- Interstitial lung disease
- Mixed restrictive and obstructive pattern pulmonary diseases
- Sleep-disordered breathing
- Alveolar hypoventilation disorders
- Chronic exposure to high altitude
- Developmental abnormalities
WHO Group IV – chronic arterial obstruction
- Chronic thromboembolic pulmonary hypertension (CTEPH)
- Other pulmonary artery obstructions
- Angiosarcoma or other tumor within the blood vessels
- Arteritis
- Congenital pulmonary artery stenosis
- Parasitic infection (hydatidosis)
WHO Group V – Pulmonary hypertension with unclear or multifactorial mechanisms
- Hematologic diseases: chronic hemolytic anemia (including sickle cell disease)
- Systemic diseases: sarcoidosis, pulmonary Langerhans cell histiocytosis: lymphangioleiomyomatosis, neurofibromatosis, vasculitis
- Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid diseases
- Others: pulmonary tumoral thrombotic microangiopathy, fibrosing mediastinitis, chronic kidney failure, segmental pulmonary hypertension (pulmonary hypertension restricted to one or more lobes of the lungs)
Genetics
Mutations in several genes have been associated with this condition these include bone morphogenetic protein receptor type 2 (BMPR2) and eukaryotic translation initiation factor 2 alpha kinase 4 gene (EIF2AK4).
Pathogenesis

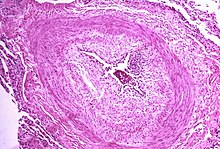
The pathogenesis of pulmonary arterial hypertension (WHO Group I) involves the narrowing of blood vessels connected to and within the lungs. This makes it harder for the heart to pump blood through the lungs, as it is much harder to make water flow through a narrow pipe as opposed to a wide one. Over time, the affected blood vessels become stiffer and thicker, in a process known as fibrosis. The mechanisms involved in this narrowing process include vasoconstriction, thrombosis, and vascular remodeling (excessive cellular proliferation, fibrosis, and reduced apoptosis/programmed cell death in the vessel walls, caused by inflammation, disordered metabolism and dysregulation of certain growth factors). This further increases the blood pressure within the lungs and impairs their blood flow. In common with other types of pulmonary hypertension, these changes result in an increased workload for the right side of the heart. The right ventricle is normally part of a low pressure system, with systolic ventricular pressures that are lower than those that the left ventricle normally encounters. As such, the right ventricle cannot cope as well with higher pressures, and although right ventricular adaptations (hypertrophy and increased contractility of the heart muscle) initially help to preserve stroke volume, ultimately these compensatory mechanisms are insufficient; the right ventricular muscle cannot get enough oxygen to meet its needs and right heart failure follows. As the blood flowing through the lungs decreases, the left side of the heart receives less blood. This blood may also carry less oxygen than normal. Therefore, it becomes harder and harder for the left side of the heart to pump to supply sufficient oxygen to the rest of the body, especially during physical activity.
In PVOD (WHO Group 1'), pulmonary blood vessel narrowing occurs preferentially (though not exclusively) in post-capillary venous blood vessels. PVOD shares several characteristics with PAH, but there are also some important differences, for example differences in prognosis and response to medical therapy.
Persistent pulmonary hypertension of the newborn occurs when the circulatory system of a newborn baby fails to adapt to life outside the womb; it is characterized by high resistance to blood flow through the lungs, right-to-left cardiac shunting and severe hypoxemia.
Pathogenesis in pulmonary hypertension due to left heart disease (WHO Group II) is completely different in that constriction or damage to the pulmonary blood vessels is not the issue. Instead, the left heart fails to pump blood efficiently, leading to pooling of blood in the lungs and back pressure within the pulmonary system. This causes pulmonary edema and pleural effusions. In the absence of pulmonary blood vessel narrowing, the increased back pressure is described as 'isolated post-capillary pulmonary hypertension' (older terms include 'passive' or 'proportionate' pulmonary hypertension or 'pulmonary venous hypertension'). However, in some patients, the raised pressure in the pulmonary vessels triggers a superimposed component of vessel narrowing, which further increases the workload of the right side of the heart. This is referred to as 'post-capillary pulmonary hypertension with a pre-capillary component' or 'combined post-capillary and pre-capillary pulmonary hypertension' (older terms include 'reactive' or 'out-of-proportion' pulmonary hypertension).
In pulmonary hypertension due to lung diseases and/or hypoxia (WHO Group 3), low levels of oxygen in the alveoli (due to respiratory disease or living at high altitude) cause constriction of the pulmonary arteries. This phenomenon is called hypoxic pulmonary vasoconstriction and it is initially a protective response designed to stop too much blood flowing to areas of the lung that are damaged and do not contain oxygen. When the alveolar hypoxia is widespread and prolonged, this hypoxia-mediated vasoconstriction occurs across a large portion of the pulmonary vascular bed and leads to an increase in pulmonary arterial pressure, with thickening of the pulmonary vessel walls contributing to the development of sustained pulmonary hypertension. Prolonged hypoxia also induces the transcription factor HIF1A, which directly activates downstream growth factor signaling that causes irreversible proliferation and remodeling of pulmonary arterial endothelial cells, leading to chronic pulmonary arterial hypertension.
In CTEPH (WHO Group 4), the initiating event is thought to be blockage or narrowing of the pulmonary blood vessels with unresolved blood clots; these clots can lead to increased pressure and shear stress in the rest of the pulmonary circulation, precipitating structural changes in the vessel walls (remodeling) similar to those observed in other types of severe pulmonary hypertension. This combination of vessel occlusion and vascular remodeling once again increases the resistance to blood flow and so the pressure within the system rises.
Molecular pathology
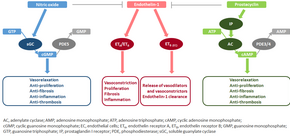
The molecular mechanism of pulmonary arterial hypertension (PAH) is not known yet, but it is believed that the endothelial dysfunction results in a decrease in the synthesis of endothelium-derived vasodilators such as nitric oxide and prostacyclin. Moreover, there is a stimulation of the synthesis of vasoconstrictors such as thromboxane and vascular endothelial growth factor (VEGF). These result in a severe vasoconstriction and vascular smooth muscle and adventitial hypertrophy characteristic of patients with PAH.
Nitric oxide-soluble guanylate cyclase pathway
In normal conditions, the vascular endothelial nitric oxide synthase produces nitric oxide from L-arginine in the presence of oxygen.
This nitric oxide diffuses into neighboring cells (including vascular smooth muscle cells and platelets), where it increases the activity of the enzyme soluble guanylate cyclase, leading to increased formation of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP). The cGMP then activates cGMP-dependent kinase or PKG (protein kinase G). Activated PKG promotes vasorelaxation (via a reduction of intracellular calcium levels), alters the expression of genes involved in smooth muscle cell contraction, migration and differentiation, and inhibits platelet activation. Nitric oxide–soluble guanylate cyclase signaling also leads to anti-inflammatory effects.
Phosphodiesterase type 5 (PDE5), which is abundant in the pulmonary tissue, hydrolyzes the cyclic bond of cGMP. Consequently, the concentration of cGMP (and thus PKG activity) decreases.
Endothelin
Endothelin-1 is a peptide (comprising 21 amino acids) that is produced in endothelial cells. It acts on the endothelin receptors ETA and ETB in various cell types including vascular smooth muscle cells and fibroblasts, leading to vasoconstriction, hypertrophy, proliferation, inflammation, and fibrosis. It also acts on ETB receptors in endothelial cells; this leads to the release of both vasoconstrictors and vasodilators from those cells, and clears endothelin-1 from the system.
Prostacyclin (and thromboxane)
Prostacyclin is synthesized from arachidonic acid in endothelial cells. In vascular smooth muscle cells, prostacyclin binds mainly to the prostaglandin I receptor. This sends a signal to increase adenylate cyclase activity, which leads to increased synthesis of cyclic adenosine monophosphate (cAMP). This in turn leads to increased cAMP-dependent protein kinase or PKA (protein kinase A) activity, ultimately promoting vasodilation and inhibiting cell proliferation. Prostacyclin signaling also leads to anti-thrombotic, anti-fibrotic, and anti-inflammatory effects. Levels of cAMP (which mediates most of the biological effects of prostacyclin) are reduced by phosphodiesterases 3 and 4. The vasoconstrictor thromboxane is also synthesized from arachidonic acid. In PAH, the balance is shifted away from synthesis of prostacyclin towards synthesis of thromboxane.
Other pathways
The three pathways described above are all targeted by currently available medical therapies for PAH. However, several other pathways have been identified that are also altered in PAH and are being investigated as potential targets for future therapies. For example, the mitochondrial enzyme pyruvate dehydrogenase kinase (PDK) is pathologically activated in PAH, causing a metabolic shift from oxidative phosphorylation to glycolysis and leading to increased cell proliferation and impaired apoptosis. Expression of vasoactive intestinal peptide, a potent vasodilator with anti-inflammatory and immune-modulatory roles, is reduced in PAH, while expression of its receptor is increased. Plasma levels of serotonin, which promotes vasoconstriction, hypertrophy and proliferation, are increased in patients with PAH, although the role played by serotonin in the pathogenesis of PAH remains uncertain. The expression or activity of several growth factors (including platelet-derived growth factor, basic fibroblast growth factor, epidermal growth factor, and vascular endothelial growth factor) is increased and contributes to vascular remodeling in PAH. Other factors underlying the proliferative state of pulmonary vascular smooth muscle cells include OPG and TRAIL. Focusing only on the pulmonary vasculature provides an incomplete picture of PAH; the ability of the right ventricle to adapt to the increased workload varies between patients and is an important determinant of survival. The molecular pathology of PAH in the right ventricle is therefore also being investigated, and recent research has shifted to consider the cardiopulmonary unit as a single system rather than two separate systems. Importantly, right ventricular remodeling is associated with increased apoptosis; this is in contrast to pulmonary vascular remodeling which involves inhibition of apoptosis.
Diagnosis



In terms of the diagnosis of pulmonary hypertension, it has five major types, and a series of tests must be performed to distinguish pulmonary arterial hypertension from venous, hypoxic, thromboembolic, or unclear multifactorial varieties. PAH is diagnosed after exclusion of other possible causes of pulmonary hypertension.
Physical examination
A physical examination is performed to look for typical signs of pulmonary hypertension (described above), and a detailed family history is established to determine whether the disease might be heritable. A history of exposure to drugs such as benfluorex (a fenfluramine derivative), dasatinib, cocaine, methamphetamine, ethanol leading to cirrhosis, and tobacco leading to emphysema is considered significant. Use of selective serotonin reuptake inhibitors during pregnancy (particularly late pregnancy) is associated with an increased risk of the baby developing persistent pulmonary hypertension of the newborn.
Echocardiography
If pulmonary hypertension is suspected based on the above assessments, echocardiography is performed as the next step. A meta-analysis of Doppler echocardiography for predicting the results of right heart catheterization reported a sensitivity and specificity of 88% and 56%, respectively. Thus, Doppler echocardiography can suggest the presence of pulmonary hypertension, but right heart catherization (described below) remains the gold standard for diagnosis of PAH. Echocardiography can also help to detect congenital heart disease as a cause of pulmonary hypertension.
28/UOTW_29_-_Ultrasound_of_the_Week_1.webm/120px--UOTW_29_-_Ultrasound_of_the_Week_1.webm.jpg">Play media
4 month old with pulmonary hypertension as seen on ultrasound
 Play media
Play media4 month old with pulmonary hypertension as seen on ultrasound
 Play media
Play media4 month old with pulmonary hypertension as seen on ultrasound

4 month old with pulmonary hypertension as seen on ultrasound
 Play media
Play mediaLong standing pulmonary hypertension
 28/UOTW_29_-_Ultrasound_of_the_Week_1.webm/120px--UOTW_29_-_Ultrasound_of_the_Week_1.webm.jpg">
28/UOTW_29_-_Ultrasound_of_the_Week_1.webm/120px--UOTW_29_-_Ultrasound_of_the_Week_1.webm.jpg">



Exclude other diseases
If the echocardiogram is compatible with a diagnosis of pulmonary hypertension, common causes of pulmonary hypertension (left heart disease and lung disease) are considered and further tests are performed accordingly. These tests generally include electrocardiography (ECG), pulmonary function tests including lung diffusion capacity for carbon monoxide and arterial blood gas measurements, X-rays of the chest and high-resolution computed tomography (CT) scanning.
Ventilation/perfusion scintigraphy
If heart disease and lung disease have been excluded, a ventilation/perfusion scan is performed to rule out CTEPH. If unmatched perfusion defects are found, further evaluation by CT pulmonary angiography, right heart catheterization, and selective pulmonary angiography is performed.
CT scan

Signs of pulmonary hypertension on CT scan of the chest are:
- Enlargement of the pulmonary trunk (measured at its bifurcation). It is, however, a poor predictor of pulmonary hypertension in patients with interstitial lung disease.
- A diameter of more than 27 mm for women and 29 mm for men is suggested as a cutoff.
- A cutoff of 31.6 mm may be a more statistically robust in individuals without interstitial lung disease.
- Increased ratio of the diameter of the main pulmonary artery (pulmonary trunk) to the ascending aorta (measured at its bifurcation).
- A ratio of 1.0 is suggested as a cutoff in adults.
- Cutoff ~1.09 in children.
- Increased diameter ratio of segmental arteries to bronchi. This finding in three or four lobes, in the presence of a dilated pulmonary trunk (≥29 mm), and absence of significant structural lung disease confers a specificity of 100% for pulmonary hypertension.
- Mural calcification in central pulmonary arteries is most frequently seen in patients with Eisenmenger's syndrome.
Right heart catheterization
Although pulmonary arterial pressure (PAP) can be estimated on the basis of echocardiography, pressure measurements with a Swan-Ganz catheter inserted through the right side of the heart provide the most definite assessment.[42] Pulmonary hypertension is defined as a mean PAP of at least 20 mm Hg (3300 Pa) at rest, and PAH is defined as precapillary pulmonary hypertension (i.e. mean PAP ≥ 20 mm Hg with pulmonary arterial occlusion pressure [PAOP] ≤ 15 mm Hg and pulmonary vascular resistance [PVR] > 3 Wood Units). PAOP and PVR cannot be measured directly with echocardiography. Therefore, diagnosis of PAH requires right-sided cardiac catheterization. A Swan-Ganz catheter can also measure the cardiac output; this can be used to calculate the cardiac index, which is far more important in measuring disease severity than the pulmonary arterial pressure. Mean PAP (mPAP) should not be confused with systolic PAP (sPAP), which is often reported on echocardiogram reports. A systolic pressure of 40 mm Hg typically implies a mean pressure of more than 25 mm Hg. Roughly, mPAP = 0.61•sPAP + 2.
Other
For people considered likely to have PAH based on the above tests, the specific associated condition is then determined based on the physical examination, medical/family history and further specific diagnostic tests (for example, serological tests to detect underlying connective tissue disease, HIV infection or hepatitis, ultrasonography to confirm the presence of portal hypertension, echocardiography/cardiac magnetic resonance imaging for congenital heart disease, laboratory tests for schistosomiasis, and high resolution CT for PVOD and pulmonary capillary hemangiomatosis). Routine lung biopsy is discouraged in patients with PAH, because of the risk to the patient and because the findings are unlikely to alter the diagnosis and treatment.
Treatment
Treatment of pulmonary hypertension is determined by whether the PH is arterial, venous, hypoxic, thromboembolic, or miscellaneous. If it is caused by left heart disease, the treatment is to optimize left ventricular function by the use of medication or to repair/replace the mitral valve or aortic valve. Patients with left heart failure or hypoxemic lung diseases (groups II or III pulmonary hypertension) should not routinely be treated with vasoactive agents including prostanoids, phosphodiesterase inhibitors, or endothelin antagonists, as these are approved for the different condition called primary pulmonary arterial hypertension. To make the distinction, doctors at a minimum will conduct cardiac catheterization of the right heart, echocardiography, chest CT, a six-minute walk test, and pulmonary function testing. Using treatments for other kinds of pulmonary hypertension in patients with these conditions can harm the patient and wastes substantial medical resources.
High dose calcium channel blockers are useful in only 5% of IPAH patients who are vasoreactive by Swan-Ganz catheter. Unfortunately, calcium channel blockers have been largely misused, being prescribed to many patients with non-vasoreactive PAH, leading to excess morbidity and mortality. The criteria for vasoreactivity have changed. Only those patients whose mean pulmonary artery pressure falls by more than 10 mm Hg to less than 40 mm Hg with an unchanged or increased cardiac output when challenged with adenosine, epoprostenol, or nitric oxide are considered vasoreactive. Of these, only half of the patients are responsive to calcium channel blockers in the long term.
A number of agents have recently been introduced for primary and secondary PAH. The trials supporting the use of these agents have been relatively small, and the only measure consistently used to compare their effectivity is the "6 minute walk test". Many have no data on mortality benefit or time to progression.
Vasoactive substances
Many pathways are involved in the abnormal proliferation and contraction of the smooth muscle cells of the pulmonary arteries in patients with pulmonary arterial hypertension. Three of these pathways are important since they have been targeted with drugs — endothelin receptor antagonists, phosphodiesterase type 5 (PDE-5) inhibitors, and prostacyclin derivatives