Cardiac Amyloidosis

Cardiac amyloidosis is a subcategory of amyloidosis where there is depositing of the protein amyloid in the cardiac muscle and surrounding tissues. Amyloid, a misfolded and insoluble protein, can become a deposit in the heart’s atria, valves, or ventricles. These deposits can cause thickening of different sections of the heart, leading to decreased cardiac function. The overall decrease in cardiac function leads to a plethora of symptoms. This multisystem disease was often misdiagnosed, with diagnosis previously occurring after death during autopsy. However, recent advancements of technologies have increased the diagnosis of the disease. Cardiac amyloidosis has multiple sub-types including light chain, familial, and senile. One of the most studied types is light chain cardiac amyloidosis. Prognosis depends on the extent of the deposits in the body and the type of amyloidosis. New treatment methods are actively being researched in regards to the treatment of heart failure and specific cardiac amyloidosis problems.
Types
The multiple subtypes of cardiac amyloidosis have varying epidemiological, diagnostic, and prognostic characteristics.
Light chain (AL-CM)
This relatively rare form of cardiac amyloidosis occurs in an estimated 6-10 cases per 1,000,000 people. This sub- type usually affects males over the age of 60 and is rapidly progressive. Pathogenesis of this form is due to the aggregation of immunoglobulin lambda light chains. These chains are created by an abnormal expansion of plasma cells. Over time, these light chains deposit into the interstitial tissue within the myocardium. Diagnostic tests includes serum and urine electrophoresis, laboratory testing for the determination of elevated levels of troponin and BNP, and ECGs showing low QRS voltages.
Familial (ATTRm-CM)
This type is caused by mutations of proteins involved in amyloid formation, including transthyretin (TTR), fibrinogen, apolipoprotein A1, or apolipoprotein A2. Due to the multiple number of potential genetic causes the incidence of this form is variable. The vast majority of familial cardiac amyloidosis still present after the age of 60. A common mutation is the TTR gene mutation Val122Ile. It is estimated that 3.5 - 4% of African Americans in The United States have the Val 122lle mutation. This type of amyloidosis can be identified by genetic testing for protein mutation. For the diagnosis of familial cardiac amyloidosis to be made a biopsy with histological evaluation must be obtained. In this histological evaluation special stains are utilized to visualize the amyloid deposits. One such stain is Congo Red, which binds specifically to the amyloid deposit and can be characterized by various lighting methods. Under polarized light, the amyloid deposits while show pathognomonic apple green birefringence, and under plain light the deposits will appear a light salmon pink color. Familial amyloidosis symptoms are centered around neuropathological and cardiac problems. Cardiac manifestations of the TTR mutation present more often in The United States.
Senile (ATTRwt-CM)
This type is considered the wild-type mutation which leads to the development of TTR deposits. It usually affects males over 70 years with the manifestation of carpal tunnel syndrome. Similar to the other subtypes of cardiac amyloidosis, a biopsy is required for diagnosis. However, formal diagnosis of Senile cardiac amyloidosis is a diagnosis of exclusion. Biopsy with histological evaluation can rule out Light chain and Familial subtypes, leaving the diagnosis of Senile. This type is often misdiagnosed, however, greater use of cardiac magnetic resonance has increased the rate of diagnosis The severity of the disease tends to be less than the Light chain and Familial variants. This is due to the amount of time that it takes to accumulate the amyloid depositions being longer in the Senile variant.
Cause
The general cause of cardiac amyloidosis is the misfolding of a specific protein precursor depending on the amyloidosis type. Protein precursors include immunoglobulin-derived light chains and transthyretin mutations. The misfolding of the protein causes it to have insoluble beta-pleated sheets, creating an amyloid. Amyloid, the aggregation, or clumping, of proteins, is resistant to degradation by the body. Amyloids are mostly fibrils, while also containing a P component, apolipoprotein, collagen, fibronectin, and laminin. The P component, a pentameric protein, stabilizes the fibrils of the amyloid, which reduces their clearance from the body. Deposits of the amyloids can occur throughout the body, including the heart, liver, kidneys, spleen, adrenal glands, and bones. Deposits in the extracellular cardiac space can stiffen the heart, resulting in restriction of the ventricles. This restriction in ventricular motion results in a decreased ability for the heart to pump efficiently, leading to the various symptoms associated with cardiac amyloidosis.
Symptoms
Symptoms of cardiac amyloidosis are a combination of heart failure and amyloid deposition in various other organs. Amyloid deposition in the heart causes restrictive diastolic heart failure that progresses to systolic heart failure.
Cardiac manifestations include:
- Dyspnea on exertion
- Peripheral edema and ascites
- Pericardial effusion
- Arrhythmias (secondary to disruption of the normal electrical system of the heart)
- Atrial arrhythmias (such as atrial fibrillation)
- First/second degree heart blocks
- Syncope
- Elevated neck veins and jugular venous pressure
- Myocardial ischemia/Angina (secondary to amyloid deposition in the small arteries of the heart)
- Myocardial oxygen demand is increased in patients with cardiac amyloidosis, regardless of changes to coronary perfusion.
For patients with light-chain amyloidosis, there can be depositions of amyloid into numerous different organs. Deposition of amyloid into other organs makes the diagnosis of cardiac amyloidosis difficult as these extracardiac manifestations mask the diagnosis. Extracardiac manifestations include:
- Macroglossia
- Periorbital bruising
- Loss of the third and fourth heart sound
- Thromboembolisms
- Symmetric sensory neuropathy (such as bilateral carpal tunnel)
- Postural hypotension (secondary to autonomic neuropathy)
- Nephrotic syndrome (secondary to free light chain damage to the kidneys/deposition of amyloid in the kidneys)
Diagnosis
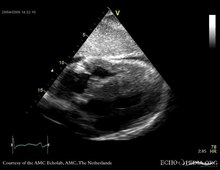 Play media
Play mediaEchocardiography
Echocardiography is a safe and non-invasive method that can be used to assess the structural and functional disease of the heart. Amyloidosis presents with ventricle and valvular thickening, biatrial enlargement, restrictive filling pattern, with normal to mildly reduced systolic function and decreased diastolic filling. An echo can be used to evaluate for prognosis of the disease, measuring the different strains within the hear. Cardiac amyloidosis produces specific alterations to the functionality of the heart. Echocardiography can be utilized to detect this specific pattern (relative preservation of the apical myocardium with decreased longitudinal strain in the mid and basal sections), which is 90-95% sensitive and 80-85% specific for cardiac amyloidosis. Echocardiography can be used to help physicians with diagnosis, however, it can only be used for the suggestion of the disease, not the confirmation, unless it is late stage amyloidosis.
ECG/EKG
ECGs of patients with cardiac amyloidosis usually show a low voltage in the limb leads, with an unusual extreme right axis. There is usually a normal P-wave, however, it can be slightly prolonged. For patients with light-chain amyloidosis, the QRS complex pattern is skewed, with poor R-waves of the chest leads.
Holter ECGs can be used to identify asymptomatic arrhythmias.
EKG changes may be present, showing low voltage and conduction abnormalities like atrioventricular block or sinus node dysfunction.
Laboratory Tests
Laboratory tests including urea and creatinine levels, liver enzymes, glucose, thyroid function, full blood count, and clotting tests. The analysis of serum and urine for presence of monoclonal immunoglobulin is also done through immunofixation for detection of the monoclonal band. Presence of the monoclonal band would be consistent with light chain amyloidosis. For light chain amyloidosis, serum immunoglobulin free light chain assay can be used for diagnosis and following of the amyloidosis. In light-chain amyloidosis, a low paraprotein level can be present.
Cardiac Biomarkers
There are 2 main cardiac biomarkers used in the assessment of cardiac amyloidosis, troponin and N-terminal proBNP. As expected, with cardiac damage and dysfunction, there can be an elevation of these markers in patients with cardiac amyloidosis. These markers have been incorporated into the various staging/scoring systems used by physicians to determine severity of the disease and prognosis.

Biopsies
Extracardiac biopsies of tissues of the kidney, liver, peripheral nerve, or abdominal fat can be used to confirm the presence of amyloid deposits. Amyloid deposits in biopsy samples are confirmed through the use of Congo red dye, which produces a green birefringence when viewed under polarized light. Sirius red staining or electron microscopy examination can also be done. The determination of the type of amyloid can be done by immunohisto-labeling techniques as well as immunofluorescence staining.
For light-chain amyloidosis patients, bone marrow biopsies could be conducted to determine the baseline percentage of plasma cells and to rule out multiple myeloma.
Catherization
Right heart catheterization is the test used to test for elevated diastolic ventricular pressures. This test is more invasive and would be performed after inconclusive endomyocardial biopsy samples.
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance (CMR) is capable of measuring the thickness of different areas of the heart. This can be used for quantification of the deposits in the heart. CMR also shows the characterization of myocardial tissue through patterns of gadolinium enhancements. However, none of the CMR technique is able to differentiate ATTR-CM and AL-CM definitely.
For AL-CM, 68% of them have symmetrical and concentric left ventricular hypertrophy. On the other hand, for ATTR-CM, 79% of them have asymmetrical left ventricular hypertrophy and 18% of them have symmetrical and concentric left ventricular hypertrophy.
In T1-weighted imaging, edema in the heart can be detected with a high T1 signal. Meanwhile, enlargement of heart cells will reduce the T1 signal. Using T1 signal, Extracellular volume (ECV) is useful to determine the degree of amyloid deposition around the heart cells and detect the regression of amyloid deposits after treatment. ECV is higher in ATTR-CM than in AL-CM.
In T2-weighted imaging, the T2 signal is increased in acute myocarditis (inflammation of heart muscles), and myocardial infarction (heart attack). T2 signal is also increased in AL-CM and ATTR-CM but the signal is greater in AL-CM before starting chemotherapy.
Late gadolinum enhancement (LGE) can determine the severity of deposition of amyloid in heart tissue. The higher the LGE signal, the more severe the heart involvement. It can be divided into three stages: no LGE, subendocardial LGE, and full-thickness (transmural) LGE.
Scintigraphy/Radionuclide Imaging
Scintigraphy can be used to measure the extent and distribution of the amyloid throughout the body, including the liver, kidney, spleen, and heart. A radiolabelled serum amyloid P component can be administered to a patient intravenously and the P component pools to the amyloid deposit proportional to the size of the deposit. The labeling of the P component can then be pictured by a gamma camera.
Technetium radionuclide scans can now reliably diagnosis cardiac amyloidosis, with certain scanning methods having greater than 99% sensitivity (but only 91% specific for amyloidosis). In this method of imaging, radiolabeled technetium is injected into the body where it binds to cardiac amyloid deposits. A subsequent scan is taken to determine where the tracer stays, therefore highlighting the amyloid deposition in the heart. This method allows for a noninvasive definitive diagnosis of cardiac amyloidosis (as in the past an endomyocardial biopsy was required)
Mass Spectrometry
Mass spectrometry can be used to determine whether the protein is light-chain or familial amyloidosis by identifying the protein subunit.
Prognosis
Overall prognosis is dependent on the extent of cardiac dysfunction. Worse outcomes have been seen when echocardiography shows left ventricular wall thickness, poor systolic function and severe diastolic dysfunction.
Light chain (AL-CM) Prognosis: For light-chain amyloidosis early detection leads to best possibility of therapies prolonging the period of remission. Well treated light chain cardiac amyloidosis has a 4 year survival rate of around 90%. In patients that undergo stem cell transplant the average survival time increases to 10 years. Staging systems have been developed to stratify severity of the disease, including the Mayo Biomarker Stage, which utilizes various biomarkers such as troponin I, troponin T, BNP, and NT-proBNP, and Free light chain concentrations.
Familial (ATTRm-CM) Prognosis: Due to the extensive number of variables involved in this subtype, prognosis varies depending on the specific type of familial cardiac amyloidosis. Variables involve mutant vs wild type transthyretin mutation and age of onset of symptoms. In comparison to light chain amyloidosis, the familial subtype is slower to progress and has a more favorable prognosis. However, the Val 122lle mutation (most common cause of familial cardiac amyloidosis) has a 4 year survival rate of 16% with an average length of 26 months. A delay in recognition plays a major factor in this reduced survival rate.
Treatments
Treatments differ according to type of amyloidosis present. The majority of treatment is aimed at preserving heart function and treating heart failure symptoms.
Light chain (AL-CM) Treatment: Since the cause of this subtype of cardiac amyloidosis is the excessive production of free light chains, the major goal of treatment is the reduction in concentration of light chains. For light-chain amyloidosis, the use of FLC assays and NT-proBNP levels can be used to monitor the progression of amyloidosis and any response to treatments. One of the major routes to decrease the production of these excess light chains is to kill the abnormal cells that are producing them. Chemotherapeutic agents such as melphalan or bortezomib can be used to kill off the abnormal cell line that is producing the free light chains. Following chemotherapy, a bone marrow transplant can be utilized to restore the normal cell lines. There are newer medications (ixazomib, carfilzomib, daratumumab, elotuzumab) under research for the treatment of multiple myeloma that can help to decrease the production of free light chains. New data suggests that orthotopic heart transplant followed by melphalan and stem cell transplant produces results similar to non cardiac amyloidosis indicated heart transplant. To treat complications, medications can be prescribed including midodrine for autonomic neuropathy, amiodarone for patients with atrial fibrillation to prevent arrhythmias, and warfarin used after a cardioembolic episode. Beta-blockers should be avoided due to the usual symptom of hypotension.
Familial (ATTRm-CM) Treatment: In recent years there have been developments in the treatment of Familial/Transthyretin cardiac amyloidosis including methods to suppress transthyretin production, stabilize amyloid fibrils, and medications that can destroy already existing fibrils. For familial amyloidosis, ACE-inhibitors and beta-blockers can be prescribed if there is no autonomic neuropathy.
- Suppression of transthyretin production: liver transplantation and medications that decreases the activity of the transthyretin genes (patisiran and inotersen). In patients with familial transthyretin mutations, liver transplantation can provide the body with a source of normal transthyretin. By changing the source of transthyretin from the original liver that contains the mutated transthyretin to a healthy liver, there will be no more production of the abnormal protein. However, liver transplant does not reverse the disease. The goal of liver transplant is to prevent additional amyloid deposition and prevent new symptoms/complications from happening. These medications bind to the mRNA of transthyretin and prevent the production of the transthyretin protein, thus decreasing the overall amount of transthyretin that can accumulate in the body.
- Stabilization of abnormal transthyretin: There are medications that can stabilize the normally folded transthyretin, preventing misfolding and subsequent amyloid deposition. These medications include Tafamidis, the NSAID Diflunisal, and AG10. Tafamidis is a medication that binds to transthyretin and keeps it in its normal shape, stopping it from aggregating into amyloid fibrils. Diflunisal and AG10 work in a similar manner to Tafamidis in their ability to bind to and stabilize Transthyretin.
- Destruction of existing amyloid fibrils: There are multiple medications that show amyloid destroying properties, Doxycycline, Tauro-ursodeoxy-cholic acid (TUDCA), and monoclonal antibodies.
The use of pacemakers (both right ventricular pacing and biventricular pacing) or implantable cardioverter defibrillators remains questionable in cardiac amyloidosis.