Fibrodysplasia Ossificans Progressiva

Fibrodysplasia ossificans progressiva (FOP), also known as Münchmeyer disease, is an extremely rare connective tissue disease. It is a severe, disabling disorder with no current cure or treatment. It is the only known medical condition where one organ system changes into another.
Fibrodysplasia ossificans progressiva is caused by a mutation of the gene ACVR1. The mutation affects the body's repair mechanism, causing fibrous tissue including muscle, tendons, and ligaments to be ossified, either spontaneously or when damaged as the result of trauma. In many cases, otherwise minor injuries can cause joints to become permanently fused as new bone forms and replaces the damaged muscle tissue. This new bone formation (known as "heterotopic ossification") eventually forms a secondary skeleton and progressively restricts the patient's ability to move. Bone formed as a result of this process is identical to "normal" bone, simply in improper locations. Circumstantial evidence suggests that the disease can cause joint degradation separate from its characteristic bone growth.
Surgical removal of the extra bone growth has been shown to cause the body to "repair" the affected area with additional bone. Although the rate of bone growth may differ depending on the patient, the condition ultimately leaves those affected immobilized as new bone replaces musculature and fuses with the skeleton.
Some patients have attempted to position their bodies in a position they would prefer to be positioned in during a flare-up to improve their quality of life.
Signs and symptoms
For unknown reasons, children born with FOP often have malformed big toes, sometimes missing a joint or, in other cases, simply presenting with a notable lump at the minor joint. The first "flare-up" that leads to the formation of FOP bone usually occurs before the age of 10. The bone growth generally progresses from the top of the body downward, just as bones grow in fetuses. A child with FOP will typically develop additional bones starting at the neck, then at the shoulders, arms, chest area, and finally at the feet.
Specifically, ossification is typically first seen in the dorsal, axial, cranial and proximal regions of the body. Later the disease progresses in the ventral, appendicular, caudal and distal regions. However, it does not necessarily occur in this order due to injury-caused flare-ups. Often, the tumor-like lumps that characterize a flare-up of the disease appear suddenly.
Bone growth occurring during flare-ups may result in the loss of mobility to affected joints, including, if the jaw/mandible is involved, the inability to fully open the mouth, limiting speech and eating. A specific occurrence of a flare-up of this condition in the foot/ankle joints can result in the limited ability to put a foot flat on the ground. Bone growth can also result in the immobilization of the hip or knee, affecting the individual's ability to walk. Extra bone formation around the rib cage restricts the expansion of lungs and diaphragm causing respiratory complications.
Since the disorder is so rare, the condition may be misdiagnosed as cancer or fibrosis. This leads physicians to order biopsies which can exacerbate the growth of FOP bone. The presence of malformed toes or thumbs in those born with FOP help distinguish this disorder from other skeletal problems.
With proper medical management the median age of survival is 40 years. However, delayed diagnosis, trauma, and infections can decrease life expectancy.
Causes
FOP is caused by an autosomal dominant allele on chromosome 2q23-24. The allele has variable expressivity, but complete penetrance. Most cases are caused by spontaneous mutation in the gametes; most people with FOP cannot or choose not to have children. A similar but less catastrophic disease is fibrous dysplasia, which is caused by a post-zygotic mutation.
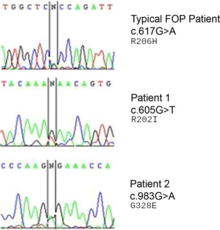
A mutation in the gene ACVR1 (also known as activin-like kinase 2 (ALK2)) is responsible for the disease. ACVR1 encodes activin receptor type-1, a BMP type-1 receptor. The mutation causes substitution of codon 206 from arginine to histidine in the ACVR1 protein. This substitution causes abnormal activation of ACVR1, leading to the transformation of connective tissue and muscle tissue into a secondary skeleton. This causes endothelial cells to transform to mesenchymal stem cells and then to bone.
Genetics
FOP is an autosomal dominant disorder. Thus, a child of an affected heterozygous parent and an unaffected parent has a 50% probability of being affected. Two affected individuals can produce unaffected children. Two unaffected individuals can produce an affected offspring as a result of the mutation of the gene. The homozygous dominant form is more severe than the heterozygous form.
The protein that causes ossification is normally deactivated by an inhibitory protein after a fetus's bones are formed in the womb, but in patients with FOP, the protein keeps working. Aberrant bone formation in patients with FOP occurs when injured connective tissue or muscle cells at the sites of injury or growth incorrectly express an enzyme for bone repair during apoptosis (self-regulated cell death), resulting in lymphocytes containing excess bone morphogenetic protein 4 (BMP4) provided during the immune system response. The bone that results occurs independently of the normal skeleton, forming its own discrete skeletal elements. These elements, however, can fuse with normal skeletal bone. The diaphragm, tongue, and extra-ocular muscles are spared in this process, as well as cardiac and smooth muscle. Since the incorrect enzyme remains unresolved within the immune response, the body continues providing the incorrect BMP4-containing lymphocytes. BMP4 is a product that contributes to the development of the skeleton in the normal embryo.
The ACVR1 gene encodes a bone morphogenic protein (BMP) receptor; this gene is mutated in FOP. It is responsible for growth and development of bone and muscles. The typical mutation, R202H, makes the inhibitor FKBP1A bind less tightly to the activation GS-loop. The end result is that ACVR1 is not effectively turned off, and an overgrowth of bone and cartilage and fusion of joints occurs. Atypical mutations involving other residues work similarly. In some cases, the receptor can end up signalling that it's active without being bound to its activating ligand.
Most of the cases of FOP were results of a new gene mutation: these people had no history of this particular disorder in their family. There are some cases where the individual has inherited the mutation from one affected parent.
Diagnosis
Outbreaks may be measurable clinically by elevated levels of alkaline phosphatase and bone-specific alkaline phosphatase. As well as removal of all tissue bone. Another telltale sign of FOP is a shortened great toe with a malformed distal first metatarsal and a missing or abnormal first phalanx and/or interphalangeal joint.
Treatment
There is no cure or approved treatment for FOP. Attempts to surgically remove bone in a FOP patient may result in explosive growth of new bone. While undergoing anesthesia, people with FOP may encounter difficulties with intubation, restrictive pulmonary disease, and changes in the electrical conduction system of the heart. Activities that increase the risk of falling or soft tissue injury should be avoided, as even minor trauma may provoke heterotopic bone formation.
Epidemiology
As of 2017, approximately 800 cases of FOP have been confirmed worldwide making FOP one of the rarest diseases known. The estimated incidence of FOP is 0.5 cases per million people and affects all ethnicities.
History
Medical reports describing individuals affected by FOP date back as far as the seventeenth century. FOP was originally called myositis ossificans progressiva and was thought to be caused by muscular inflammation (myositis) that caused bone formation. The disease was renamed by Victor A. McKusick in 1970 following the discovery that soft tissue other than muscles (e.g. ligaments) were also affected by the disease process.
The best known FOP case is that of Harry Eastlack (1933–1973). His condition began to develop at the age of ten, and by the time of his death from pneumonia in November 1973, six days before his 40th birthday, his body had completely ossified, leaving him able to move only his lips. Eastlack never met another person with FOP during his lifetime.
Eastlack donated his body to science. His skeleton is now at the Mütter Museum in Philadelphia, and has proven to be an invaluable source of information in the study of FOP. Another FOP sufferer, Carol Orzel (April 20, 1959 – February 2018), also donated her body to the museum and her skeleton was placed on exhibit there, adjacent to Eastlack's, in February 2019.
Research
Clinical trials of isotretinoin, etidronate with oral corticosteroids, and perhexiline maleate have failed to demonstrate effectiveness, though the variable course of the disease and small prevalence induces uncertainty.
A handful of pharmaceutical companies focused on rare diseases are currently in varying stages of investigation into different therapeutic approaches for FOP.
In August 2015, U.S. Food and Drug Administration (FDA) Office of Orphan Products Development granted La Jolla Pharmaceuticals orphan drug designation for two novel compounds for FOP. The compounds are small-molecule protein kinase inhibitors designed to selectively block ACVR1 (ALK2).
In August 2015, Clementia Pharmaceuticals also began the enrollment of children (ages 6 and above) into its Phase II clinical trial investigating palovarotene for the treatment of FOP. Preclinical studies demonstrated that palovarotene, a retinoic acid receptor gamma agonist, blocked abnormal bone formation in animal models by inhibition of secondary messenger systems in the BMP pathway. Clementia licensed palovarotene from Roche Pharmaceuticals, which previously evaluated the compound in more than 800 individuals including healthy volunteers and patients with chronic obstructive pulmonary disease. Palovarotene received Fast Track designation from the FDA and orphan designations for the treatment of FOP from both the FDA and the European Medicines Agency (EMA).
In September 2015, Regeneron announced new insight into the mechanism of disease involving the activation of the ACVR1 receptor by activin A. In 2016, the company initiated a phase 1 study of its activin antibody, REGN 2477, in healthy volunteers; a phase 2 trial in FOP patients was conducted in 2017.
Another potential therapeutic approach involves allele-specific RNA interference that targets mutated mRNA for degradation while preserving normal ACVR1 gene expression.
Further investigation into the mechanisms of heterotopic bone formation in FOP could aid in the development of treatments for other disorders involving extra-skeletal bone formation.
Fibro/adipogenic progenitors (FAPs) may be the disease-causing cell type responsible for activin A dependent ectopic bone formation in both the muscles and tendons of mice bearing the FOP causing ACVR1(R206H) mutation.
Clinical trials of several potential treatments are in progress as of 2019.
See also
- International FOP Association
- Osteogenesis imperfecta, a condition characterized by bones breaking unusually easily, caused by mutations in genes related to those responsible for FOP
- FOP Friends