Xeroderma Pigmentosum

Xeroderma pigmentosum (XP) is a genetic disorder in which there is a decreased ability to repair DNA damage such as that caused by ultraviolet (UV) light. Symptoms may include a severe sunburn after only a few minutes in the sun, freckling in sun exposed areas, dry skin and changes in skin pigmentation. Nervous system problems, such as hearing loss, poor coordination, loss of intellectual function and seizures, may also occur. Complications include a high risk of skin cancer, with about half having skin cancer by age 10 without preventive efforts, and cataracts. There may be a higher risk of other cancers such as brain cancers.
XP is autosomal recessive, with at least nine specific mutations able to result in the condition. Normally, the damage to DNA which occurs in skin cells from exposure to UV light is repaired by nucleotide excision repair. In people with xeroderma pigmentosum, this damage is not repaired. As more abnormalities form in DNA, cells malfunction and eventually become cancerous or die. Diagnosis is typically suspected based on symptoms and confirmed by genetic testing.
There is no cure for XP. Treatment involves completely avoiding the sun. This includes protective clothing, sunscreen and dark sunglasses when out in the sun. Retinoid creams may help decrease the risk of skin cancer. Vitamin D supplementation is generally required. If skin cancer occurs, it is treated in the usual way. The life expectancy of those with the condition is about 30 years less than normal.
The disease affects about 1 in 100,000 worldwide. By region, it affects about 1 in 370 in India, 1 in 20,000 in Japan, 1 in 250,000 people in the United States and 1 in 430,000 in Europe. It occurs equally commonly in males and females. Xeroderma pigmentosum was first described in the 1870s by Moritz Kaposi. In 1882, Kaposi coined the term xeroderma pigmentosum for the condition, referring to its characteristic dry, pigmented skin. Individuals with the disease have been referred to as "children of the night" or "moon children".
Signs and symptoms
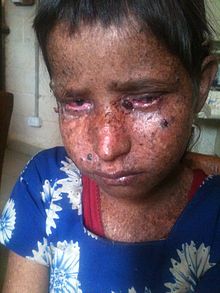
Signs and symptoms of xeroderma pigmentosum may include:
- Severe sunburn when exposed to only small amounts of sunlight. These often occur during a child's first exposure to sunlight.
- Development of many freckles at an early age
- Rough-surfaced growths (solar keratoses), and skin cancers
- Eyes that are painfully sensitive to the sun and may easily become irritated, bloodshot and clouded
- Blistering or freckling on minimum sun exposure
- Telangiectasia (spider veins)
- Limited growth of hair on chest and legs
- Scaly skin
- Xeroderma (dry skin)
- Irregular dark spots on the skin
- Corneal ulcerations
Genetics

One of the most frequent defects in xeroderma pigmentosum is an autosomal recessive genetic defect in which nucleotide excision repair (NER) enzymes are mutated, leading to a reduction in or elimination of NER. If left unchecked, damage caused by ultraviolet light can cause mutations in individual cell's DNA. The causes of the neurological abnormalities are poorly understood and are not connected with exposure to ultraviolet light. The most current theories suggest that oxidative DNA damage is generated during normal metabolism in the central nervous system, and that some types of this damage must be repaired by NER.
Since DNA repair is under genetic control, it can mutate. Many genetic disorders such as xeroderma pigmentosum (XP; MIM 278700) are caused by mutations in genes that repair damaged DNA. XP affects the mechanism that repairs UV damage in skin cell DNA. Those affected with the autosomal recessive disorder XP are extremely sensitive to UV light produced by the sun and develop pigmented spots, tumors, and skin cancer with minimal exposure. Individuals with XP are about 1,000 times more likely to develop skin cancer than individuals without the disorder.
The molecular defects in XP cells result in a greatly elevated induction of mutations in sun-exposed skin of affected individuals. This increased mutation frequency probably accounts for the pigmentation changes and the skin cancers. Examination of mutations in the p53 gene in tumors from XP patients reveal p53 mutations characteristic of UV exposure in the majority of tumors As with all genetic disorders, genetic counseling and psychological support is appropriate for the families to discuss probability of occurrence in future pregnancies, feelings of isolation and concern about career prospects. There is no cure for xeroderma pigmentosum. The most common fate for individuals with XP is early death from cancer.
XP repair proteins
The XPA protein acts during NER as a scaffold for assembly of other DNA repair proteins at sites of DNA damage to ensure appropriate excision of the damage.
The XPB (ERCC3) protein is employed in unwinding the DNA double helix after DNA damage is initially recognized. Mutations in the XPB(ERCC3) gene can lead to XP or XP combined with Cockayne syndrome.
The XPC protein forms a complex with RAD23B protein to form the initial damage recognition factor in global genomic nucleotide excision repair (GG-NER). This complex recognizes a wide variety of damages that thermodynamically destabilize DNA duplexes.
The XPD (ERCC2) protein, in combination with the XPB helicase-containing transcription/repair complex TFIIH, is employed in unwinding the DNA duplex after damage is initially recognized. Mutations in the XPD(ERCC2) gene cause a variety of syndromes; XP, trichothiodystrophy (TTD), or a combination of XP and Cockayne syndrome (XPCS). Both trichothiodystrophy and Cockayne syndrome display features of premature aging, suggesting an association between deficient DNA repair and premature aging (see DNA damage theory of aging).
XPE is a heterodimeric protein composed of two subunits. The larger subunit DDB1 primarily functions as a core component of CUL4A- and CUL4B-based E3 ubiquitin ligase complexes. Substrates that are ubiquitinnated by these complexes include proteins employed in DNA repair.
The XPF (ERCC4) protein together with the ERCC1 protein forms a complex usually designated ERCC1-XPF. This complex separates the DNA helix for a short distance on either side of the site of damage. It then acts as an endonuclease to incise the damaged DNA strand on the 5’ side of the damaged site. Mutant cells with deficient ERCC1-XPF are not only defective in NER, but also in the repair of double-strand breaks and inter-strand crosslinks.
The XPG protein is an endonuclease that incises DNA during NER at the 3’ side of the damaged nucleotide. Mutations in the XPG (ERCC5) gene can lead to XP alone, or in combination with Cockayne syndrome (CS), or in combination with infantile lethal cerebro-oculo-facio-skeletal syndrome.
Diagnosis
Types
There are seven complementation groups, plus one variant form:
| Type | Diseases Database | OMIM | Gene | Locus | Also known as / description |
| Type A, I, XPA | 29877 | 278700 | XPA | 9q22.3 | Xeroderma pigmentosum group A - the classical form of XP |
| Type B, II, XPB | 29878 | 133510 | XPB | 2q21 | Xeroderma pigmentosum group B |
| Type C, III, XPC | 29879 | 278720 | XPC | 3p25 | Xeroderma pigmentosum group C |
| Type D, IV, XPD | 29880 | 278730 278800 | XPD ERCC6 | 19q13.2-q13.3, 10q11 | Xeroderma pigmentosum group D or De Sanctis-Cacchione syndrome (can be considered a subtype of XPD) |
| Type E, V, XPE | 29881 | 278740 | DDB2 | 11p12-p11 | Xeroderma pigmentosum group E |
| Type F, VI, XPF | 29882 | 278760 | ERCC4 | 16p13.3-p13.13 | Xeroderma pigmentosum group F |
| Type G, VII, XPG | 29883 | 278780 133530 | RAD2 ERCC5 | 13q33 | Xeroderma pigmentosum group G and COFS syndrome type 3 |
| Type V, XPV | 278750 | POLH | 6p21.1-p12 | Xeroderma pigmentosum variant - these patients have mutation in a gene that codes for a specialized DNA polymerase called polymerase-η (eta). Polymerase-η can replicate over the damage and is needed when cells enter S-phase in the presence of a DNA-replication. |
Treatment

There is no cure for the disorder; all treatment is symptomatic or preventative. Symptoms can be avoided or controlled by completely avoiding exposure to sunlight, either by staying indoors or wearing protective clothing and using sunscreen when outdoors. Keratosis can also be treated by using cryotherapy or fluorouracil. In more severe cases of XP, even minuscule amounts of UV light, for example, from covered windows or fluorescent bulbs, can be very dangerous and trigger symptoms.
On September 10, 2020, Clinuvel Pharmaceuticals announced that it was investigating the use of its FDA-approved flagship drug Scenesse as a potential treatment to increase pain-free light exposure for patients with xeroderma pigmentosum.
Prognosis
The average life expectancy of an individual with any type of XP and no neurological symptoms is approximately 37 years, and 29 years if neurological symptoms are present.
In the United States, the probability for individuals with the disorder to survive until 40 years of age may be as high as 70% if they have never been exposed to sunlight in their life.
In India, many patients with XP die at an early age from skin cancers. However, if a person is diagnosed early, does not have severe neurological symptoms, and takes precautionary measures to completely avoid any exposure to UV light and sunlight, they may be able to survive until middle age.
History
Xeroderma pigmentosum was first described in 1874 by Hebra and Moritz Kaposi. In 1882, Kaposi coined the term xeroderma pigmentosum for the condition, referring to its characteristic dry, pigmented skin.
The 1968 paper about XP by James Cleaver demonstrated the link between UV-induced DNA damage, faulty DNA repair and cancer.
Culture
Because people with XP need to strictly avoid sunlight, but can go outside at night, they have been called children of the dark, children of the night, and vampire children. These terms can be considered derogatory.
XP has been a plot element in several fictional works. One of the common themes in films about XP is whether teens with XP will risk sun exposure in pursuit of a romantic partner.
Film series like Children of Darkness, a German silent-drama film which was released in two parts in the year of 1921 and 1922 respectively, was among some of the initially popular movies that was made about XP.
Other films, like the 1964 American drama film Della, starring Joan Crawford, Paul Burke, Charles Bickford and Diane Baker, directed by Robert Gist, which was originally produced by Four Star Television as a television pilot for a proposed NBC series named Royal Bay, was also based on this skin disease.
The Dark Side of the Sun, a 1988 American-Yugoslavian drama film, was directed by Božidar Nikolić and stars Brad Pitt for his first ever leading role as a young man in search of a cure for his disorder.
The Others (2001 film), a 2001 American psychological horror film starring Nicole Kidman, features two children, Anne and Nicholas, who must avoid all sunlight because of a rare disease characterized by photosensitivity.
A CBS television movie aired in 1994, Children of the Dark, was based on the story of the real-life couple Jim and Kim Harrison, whose two daughters have XP.
Lurlene McDaniel's young adult book How I Do Love Thee features the story "Night Vision", in which the protagonist, leukemia survivor Brett, falls in love with a girl named Shayla that has XP.
Christopher Snow, the protagonist of Dean Koontz's Moonlight Bay Trilogy, has XP and therefore must live most of his life during the night. The first two entries of the trilogy, Fear Nothing and Seize the Night, were both published in 1998. The final entry in the trilogy, tentatively titled Ride the Storm, has yet to be published as of August 2020.
The 2011 French drama film The Moon Child is based on a 13 year old child with XP, which prevents him from exposing himself to daylight.
The 2012 documentary Sun Kissed explores the XP problem on the Navajo Indian Reservation, and links it to the genetic legacy of the Long Walk of the Navajo, when the Navajo people were forced to move to a new location.
The 2018 romance film Midnight Sun, based on a 2006 Japanese film, A Song to the Sun, tells the story of a girl named Katie Price, who has a life-threatening sensitivity towards sunlight because of XP, and the impact of her sickness on her normal life, which eventually claims her life as she is accidentally momentarily exposed to sunlight while watching a sunrise.
Research directions
Research into XP has had two main results: better understanding the disease itself, and also better understanding the normal biological mechanisms involved in DNA repair. Research into XP has produced insights that have been translated into treatments and prevention for cancer.
See also
- DeSanctis–Cacchione syndrome
- Genetic disorder
- Biogerontology
- Cockayne syndrome
- List of cutaneous conditions
- List of cutaneous conditions associated with increased risk of nonmelanoma skin cancer
- Photophobia
- Senescence