Acral lentiginous melanoma (ALM) is a type of melanoma that occurs on the palms of the hands or the soles of the feet. The condition can develop in normal-appearing skin or within an existing mole. ALM begins as a flat patch of discolored skin that may enlarge slowly over time. Although the cancer cells often remain contained at the skins surface (epidermis) initially, ALM can become invasive and spread as the condition advances. Like other flat forms of melanoma, it can be recognized by the ABCDE rule .
Dens evaginatus involves an outfolding of the enamel organ in such a way that the occlusal surface of the affected posterior tooth has a tuberculated appearance. When these evaginations are fractured off, pulpal exposure may result. Few familial cases have been reported. However, a genetic basis was supported by Bixler (1976) on the following grounds: 1) the anomaly has been found almost only in persons of Mongoloid ancestry, although it has been observed in all parts of the world, and 2) the prevalence in persons of mixed Mongoloid ancestry is lower than in 'pure' groups. Stewart et al. (1978) observed dens evaginatus in several members of a family of Guatemalan Indian descent. Father and 2 daughters were affected. Inheritance - Autosomal dominant Teeth - Tuberculated occlusal tooth surface ▲ Close
This leads to a clinically important higher ratio of androstenedione to testosterone. [11] [3] [12] Diagnosis [ edit ] In terms of the diagnosis of 17β-hydroxysteroid dehydrogenase III deficiency the following should be taken into account: [4] [7] Increased androstenedione:testosterone ratio Thyroid dyshormonogenesis Genetic testing Management [ edit ] Further information: Intersex medical interventions The 2006 Consensus statement on the management of intersex disorders states that individuals with 17β-hydroxysteroid dehydrogenase III deficiency have an intermediate risk of germ cell malignancy, at 28%, recommending that gonads be monitored. [5] A 2010 review put the risk of germ cell tumors at 17%. [13] The management of 17β-hydroxysteroid dehydrogenase III deficiency can consist, according to one source, of the elimination of gonads prior to puberty , in turn halting masculinization . [7] Hewitt and Warne state that, children with 17β-hydroxysteroid dehydrogenase III deficiency who are raised as girls often later identify as male, describing a "well known, spontaneous change of gender identity from female to male" that "occurs after the onset of puberty." [14] A 2005 systematic review of gender role change identified the rate of gender role change as occurring in 39–64% of individuals with 17β-hydroxysteroid dehydrogenase III deficiency raised as girls. [15] Society and culture [ edit ] Modification of children's sex characteristics to meet social and medical norms is strongly contested, with numerous statements by civil society organizations and human rights institutions condemning such interventions, including describing them as "harmful practices". [16] [17] [18] Re: Carla (Medical procedure) [ edit ] Further information: Intersex rights in Australia A 2016 case before the Family Court of Australia [19] was widely reported in national, [20] [21] [22] and international media. [23] The judge ruled that parents were able to authorize the sterilization of their 5-year-old child.
A number sign (#) is used with this entry because 17-beta hydroxysteroid dehydrogenase III deficiency, resulting in male pseudohermaphroditism or polycystic ovary disease, is caused by homozygous or compound heterozygous mutation in the HSD17B3 gene (605573) on chromosome 9q22. Description HSD17B3 deficiency is an autosomal recessive disorder that manifests, in males, as undermasculinization characterized by hypoplastic-to-normal internal genitalia (epididymis, vas deferens, seminal vesicles, and ejaculatory ducts) but female external genitalia and the absence of a prostate. This phenotype is caused by inadequate testicular synthesis of testosterone, which, in turn, results in insufficient formation of dihydrotestosterone in the anlage of the external genitalia and prostate during fetal development. At the expected time of puberty, there is a marked increase in plasma leuteinizing hormone and, consequently, in testicular secretion of androstenedione. Hence, a diagnostic hallmark of this disorder is a decreased plasma testosterone-to-androstenedione ratio.
17-beta-hydroxysteroid dehydrogenase isozyme 3 (17betaHSD III) deficiency is a rare disorder leading to male pseudohermaphroditism (MPH), a condition characterized by incomplete differentiation of the male genitalia in 46X,Y males. Epidemiology The estimated incidence of this disease is 1 in 147 000 in The Netherlands. Clinical description The 17betaHSD III enzyme catalyzes the conversion of androstenedione to testosterone in the testis. Lack of testosterone in the fetal testis leads to genetic males with female external genitalia. Patients usually present at birth with female or ambiguous external genitalia, characterized by clitoromegaly, posterior labioscrotal fusion and perineal blind vaginal pouch.
17-beta hydroxysteroid dehydrogenase 3 deficiency is an inherited condition that affects male sexual development. People with this condition are genetically male and have testes, but do not produce enough testosterone . Most people with this condition are born with external genitalia that appear female. In some cases, the external genitalia are ambiguous or appear male but are abnormal in size and/or appearance. During puberty, people with this condition typically go on to develop male secondary sex characteristics, such as increased muscle mass, deepening of the voice, and development of male pattern body hair. 17-beta hydroxysteroid dehydrogenase 3 deficiency is caused by mutations in the HSD17B3 gene and is inherited in an autosomal recessive pattern.
17-beta hydroxysteroid dehydrogenase 3 deficiency is a condition that affects male sexual development. People with this condition are genetically male, with one X and one Y chromosome in each cell, and they have male gonads (testes ). Their bodies, however, do not produce enough of a male sex hormone (androgen) called testosterone. Testosterone has a critical role in male sexual development, and a shortage of this hormone disrupts the formation of the external sex organs before birth. Most people with 17-beta hydroxysteroid dehydrogenase 3 deficiency are born with external genitalia that appear female.
type of generalized seizure This article includes a list of general references , but it remains largely unverified because it lacks sufficient corresponding inline citations . Please help to improve this article by introducing more precise citations. ( August 2016 ) ( Learn how and when to remove this template message ) Absence seizure Other names Petit mal seizures Specialty Neurology Absence seizures are one of several kinds of generalized seizures . These seizures are sometimes referred to as petit mal seizures (from the French for "little illness", a term dating from the late 18th century). [1] Absence seizures are characterized by a brief loss and return of consciousness, generally not followed by a period of lethargy (i.e. without a notable postictal state ). Contents 1 Signs and symptoms 2 Risk factors 3 Diagnosis 3.1 Syndromes 4 Treatment 4.1 Medications that should not be used 4.2 Data limitations 5 References 6 External links Signs and symptoms [ edit ] The clinical manifestations of absence seizures vary significantly among patients. [2] [3] [4] Impairment of consciousness is the essential symptom, and may be the only clinical symptom, but this can be combined with other manifestations. The hallmark of the absence seizures is abrupt and sudden-onset impairment of consciousness, interruption of ongoing activities, a blank stare, possibly a brief upward rotation of the eyes.
Molecular Genetics Kalachikov et al. (2002) constructed a complete, 4.2-Mb physical map across the genetically implicated disease-gene region, identified 28 putative genes, and resequenced all or part of 21 genes before identifying presumptive mutations in 1 copy of the leucine-rich, glioma-inactivated-1 gene (604619.0001-604619.0003) in each of 5 families with ADPEAF.
A rare, genetic, familial partial epilepsy disease characterized by focal seizures associated with prominent ictal auditory symptoms, and/or receptive aphasia, presenting in two or more family members and having a relatively benign evolution. Epidemiology The prevalence of autosomal dominant epilepsy with auditory features (ADEAF) is unknown but likely to be very low. Fewer than 3% of persons with epilepsy have a significant family history of epilepsy and only a fraction of these have clinical features consistent with ADEAF. Clinical description ADEAF usually presents in adolescence or early adulthood (but can range from 4-50 years of age) with the onset of focal epilepsy originating predominantly in the lateral temporal lobe. Seizures may be precipitated by specific sounds (i.e. telephone ringing or speech) but in most cases there are no recognizable triggers.
However, two family members with epilepsy died suddenly in their sleep, both at age 28 years; a relationship to seizures was suspected but could not be confirmed [Brodtkorb et al 2002].
Autosomal dominant partial epilepsy with auditory features (ADPEAF) is an uncommon form of epilepsy that runs in families. This disorder causes seizures usually characterized by sound-related (auditory) symptoms such as buzzing, humming, or ringing. Some people experience more complex sounds during a seizure, such as specific voices or music, or changes in the volume of sounds. Some people with ADPEAF suddenly become unable to understand language before losing consciousness during a seizure. This inability to understand speech is known as receptive aphasia. Less commonly, seizures may cause visual hallucinations, a disturbance in the sense of smell, a feeling of dizziness or spinning (vertigo), or other symptoms affecting the senses.
A number sign (#) is used with this entry because of evidence that familial temporal lobe epilepsy-8 (ETL8) is caused by heterozygous mutation in the GAL gene (137035) on chromosome 11q13. One such family has been reported. For a discussion of genetic heterogeneity of temporal lobe epilepsy, see ETL1 (600512). Clinical Features Guipponi et al. (2015) reported a pair of monozygotic twin brothers who developed temporal lobe epilepsy at age 13 years. Both reported aura variably characterized by abdominal discomfort, incoherent speech, blurred vision, auditory hallucinations, slow ideation, or deja vu. The symptoms were consistent with complex partial seizures with occasional secondary generalization.
Autosomal dominant partial epilepsy with auditory features (ADPEAF) is a rare form of epilepsy , a condition that is characterized by recurrent seizures. In ADPEAF, specifically, most affected people experience secondary generalized seizures and partial seizures , some of which are associated with sound-related symptoms (such as buzzing, humming, or ringing) and/or receptive aphasia (inability to understand written or spoken words). Less commonly, seizures may cause visual hallucinations, a disturbance in the sense of smell, vertigo , or other symptoms affecting the senses. Signs and symptoms of the condition generally begin in adolescence or early adulthood. ADPEAF is caused by changes (mutations) in the LGI1 or RELN gene and is inherited in an autosomal dominant manner.
A number sign (#) is used with this entry because of evidence that familial temporal lobe epilepsy-7 (ETL7) is caused by heterozygous mutation in the RELN gene (600514) on chromosome 7q22. Description Familial temporal lobe epilepsy-7 is a form of autosomal dominant lateral temporal lobe epilepsy characterized by focal seizures with prominent auditory symptoms (summary by Dazzo et al., 2015). For a general description and a discussion of genetic heterogeneity of familial temporal lobe epilepsy, see 600512. Clinical Features Dazzo et al. (2015) reported 7 unrelated families with autosomal dominant lateral temporal lobe epilepsy. Affected individuals had focal epilepsy with auras consistent with a lateral temporal origin; auras were mainly auditory and manifest as ringing, humming, sounds, voices, or music.
Christiano et al. (1996) reported 2 unrelated families with autosomal dominant DEB. In 1 family, the proband was a 28-year-old woman who was noted to have blisters at birth.
Generalized dominant dystrophic epidermolysis bullosa (DDEB-gen) is a subtype of dystrophic epidermolysis bullosa (DEB, see this term), formerly known as DDEB, Pasini and Cockayne-Touraine types, characterized by generalized blistering, milia formation, atrophic scarring, and dystrophic nails. Epidemiology The differential diagnosis includes other forms of EB. In the neonatal period, aplasia cutis congenita, herpes simplex infection, congenital erosive and vesicular dermatosis, epidermolytic ichthyosis, linear IgA bullous dermatosis, bullous pemphigoid, neonatal pemphigus and pemphigoid gestationis, bullous impetigo, and staphylococcal scalded skin syndrome (see these terms) may need to be considered. Clinical description The clinical picture is milder than that of the autosomal recessive generalized EB forms. DDEB-gen manifests usually at birth with the development of blisters, primarily affecting the limbs. Aplasia cutis congenita (congenital absence of the skin) can also be observed, more frequently on the lower part of the legs.
Dystrophic epidermolysis bullosa is one of the major forms of a group of conditions called epidermolysis bullosa. Epidermolysis bullosa cause the skin to be very fragile and to blister easily. Blisters and skin erosions form in response to minor injury or friction, such as rubbing or scratching. The signs and symptoms of dystrophic epidermolysis bullosa vary widely among affected individuals. In mild cases, blistering may primarily affect the hands, feet, knees, and elbows.
Dominant dystrophic epidermolysis bullosa (DDEB) is a type of epidermolysis bullosa (EB), which is a group of rare inherited conditions in which the skin blisters extremely easily. DDEB is one of the milder forms of EB, although the severity is variable. Blisters may be present at birth, but typically appear during early childhood; occasionally they do not develop until later in life. Blisters often become more numerous and tend to occur over vulnerable sites such as knees, ankles, elbows and knuckles. In adulthood, they usually become less frequent and scars fade. Other signs and symptoms of DDEB may include dystrophic or absent nails, constipation, dental caries and swallowing problems.
. ^ CDC Advisory Archived 2007-05-31 at the Wayback Machine ^ "What is onchocerciasis?" . CDC . Retrieved 2010-06-28 . transmission is most intense in remote African rural agricultural villages, located near rapidly flowing streams...
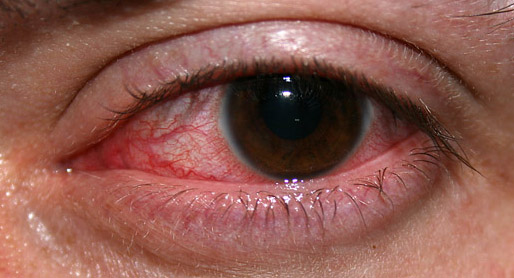
Overview Keratitis is an inflammation of the cornea — the clear, dome-shaped tissue on the front of your eye that covers the pupil and iris. Keratitis may or may not be associated with an infection. Noninfectious keratitis can be caused by a relatively minor injury, such as from wearing your contact lenses too long or getting a foreign body in the eye. Infectious keratitis can be caused by bacteria, viruses, fungi and parasites. If you have eye redness or other symptoms of keratitis, make an appointment to see an eye specialist. With prompt attention, mild to moderate cases of keratitis can usually be effectively treated without loss of vision.
"Congenital brachioesophagus with secondary intrathoratic stomach associated with rachischisis described as "serpentine-like syndrome": a case report and literature review". Pediatric Surgery International . 28 (1): 63–66. doi : 10.1007/s00383-011-3000-7 .
Van Goethem et al. (1997) reported 2 Belgian families in which 5 individuals had arPEO. Age at onset ranged from 28 to 61 years. The most prominent features were ptosis, ophthalmoplegia, generalized muscle weakness, including the neck and facial muscles, increased serum creatine kinase, and areflexia.
A rare genetic, neuro-ophthalmological disease characterized by progressive weakness of the external eye muscles, resulting in bilateral ptosis and diffuse, symmetric ophthalmoparesis. Additional signs may include generalized skeletal muscle weakness, muscle atrophy, sensory axonal neuropathy, ataxia, cardiomyopathy, and psychiatric symptoms. It is usually more severe than autosomal dominant form.
A number sign (#) is used with this entry because of evidence that autosomal recessive progressive external ophthalmoplegia with mitochondrial DNA deletions-3 (PEOB3) is caused by compound heterozygous mutation in the mitochondrial thymidine kinase gene (TK2; 188250) on chromosome 16q21. One such family has been reported. For a discussion of genetic heterogeneity of autosomal recessive PEO, see PEOB1 (258450). Clinical Features Tyynismaa et al. (2012) reported 2 Finnish sisters, born of unrelated parents, with adult-onset progressive external ophthalmoplegia beginning in their forties. Both also developed progressive proximal muscle weakness associated with muscle atrophy. One had scapular winging and 1 developed dysarthria; both patients reported dysphagia.
A disturbance in this separation process at the final closure due to the lack of apoptosis is considered to be a critical aspect of nasofrontal and nasoethmoidal encephalocele. [7] Prevention [ edit ] It is recommended that women take a multivitamin with 400 micrograms of folic acid daily to reduce the likelihood of any type of neural tube defects before and during the first 28 days after conception. [8] Treatment [ edit ] A series of steps involved in reconstructive surgery of a frontal encephalocele.
Encephaloceles are rare neural tube defects characterized by sac-like protrusions of the brain and the membranes that cover it through openings in the skull. These defects are caused by failure of the neural tube to close completely during fetal development. The result is a groove down the midline of the upper part of the skull, or the area between the forehead and nose, or the back of the skull. When located in the back of the skull, encephaloceles are often associated with neurological problems. Encephaloceles are usually dramatic deformities diagnosed immediately after birth; but occasionally a small encephalocele in the nasal and forehead region can go undetected.
A rare, neural tube closure defect characterized by partial lacking of bone fusion, resulting in sac-like protrusions of the brain and the membranes that cover it through the openings in the skull. Protruding tissue may be located on any part of the head, but most often affects the occipital area. Depending in the size nad location, encephalocele are often associated with neurological problems including intellectual disability, seizures, vision impairment, ataxia, and hydrocephalus.
Natal teeth are teeth present at birth and neonatal teeth are teeth that erupt within a month after delivery. Sibert and Porteous (1974) observed natal teeth in 6 members of a kindred. Limrick (1893) first observed natal teeth in a mother, her son and her sister's daughter. Bodenhoff and Gorlin (1963) reported a frequency of about 1 in 3,000 live births for natal teeth. Alaluusua et al. (2002) reported a prevalence of 1 in 1,000 for natal and neonatal teeth in Finland.
Léri-Weill dyschondrosteosis is a disorder of bone growth. Affected individuals typically have shortening of the long bones in the arms and legs (mesomelia). As a result of the shortened leg bones, people with Leri-Weill dyschondrosteosis typically have short stature. Most people with the condition also have an abnormality of the wrist and forearm bones called Madelung deformity, which may cause pain and limit wrist movement. This abnormality usually appears in childhood or early adolescence. Other features of Léri-Weill dyschondrosteosis can include increased muscle mass (muscle hypertrophy); bowing of a bone in the lower leg called the tibia; a greater-than-normal angling of the elbow away from the body (increased carrying angle); and a high arched palate. Léri-Weill dyschondrosteosis occurs in both males and females, although its signs and symptoms tend to be more severe in females.
Madelung deformity (MD) is a rare congenital (present from birth) condition in which the wrist grows abnormally and part of the radius, one of the bones of the forearms, stops growing early and is short and bowed. The other forearm bone, the ulna, keeps growing and can dislocate, forming a bump. Symptoms typically develop in mid- to late-childhood or early adolescence (around 6 to 13 years of age) and usually affect both wrists. It is more commonly observed in females. Symptoms include a decreased range of motion in the wrist, pain, and a visible difference in the appearance of the wrist. In addition to the abnormal growth, there is also an abnormal palmar (Vickers’ ligament) that is thought to contribute to the deformity.
"Identification and computationally-based structural interpretation of naturally occurring variants of human protein C". Hum Mutat . 28 (4): 345–55. doi : 10.1002/humu.20445 .
Congenital protein C deficiency is an inherited coagulation disorder characterized by deep venous thrombosis symptoms due to reduced synthesis and/or activity levels of protein C. Epidemiology Prevalence of severe protein C deficiency (homozygous or compound heterozygous forms) is estimated at 1/ 500,000. Partial deficiencies (heterozygous forms) are much more frequent (1/200-1/500). Men and women are equally affected. Clinical description Patients with undetectable protein C levels usually manifest the disease several hours to days after birth, with purpura fulminans (see this term) or massive venous thrombosis. Purpura fulminans is a life-threatening condition involving severe clotting throughout the body and causing necrosis of tissues.
Six cases were diagnosed by prenatal ultrasound and the pregnancies were terminated; of the 6 other cases, one died at 3 months of age of respiratory illness, but the other 5 were still alive and were aged 28 months to 15 years, demonstrating that patients with this disease can survive well beyond the neonatal period.
Opsismodysplasia is a skeletal dysplasia characterized by congenital dwarfism and facial dysmorphism. Epidemiology Twenty five cases (16 males and 9 females) have been reported in the literature so far. Clinical description The disorder is evident at birth. The facial dysmorphism includes macrocephaly, prominent brow, large fontanels, depressed nasal bridge, a small nose, anteverted nares and a long philtrum, hypertelorism, and exophthalmos. Additional features include a short neck, narrow thorax, predominantly rhizomelic micromelia with very short long bones, short feet and hands, muscular hypotonia, severe platyspondyly, marked delay of bone maturation, and increased susceptibility to respiratory infections. Etiology Etiology remains unknown. Genetic counseling Autosomal recessive transmission has been suggested.
Opsismodysplasia Other names OPSMD [1] Specialty Orthopedic Opsismodysplasia is a type of skeletal dysplasia (a bone disease that interferes with bone development) first described by Zonana and associates in 1977, and designated under its current name by Maroteaux (1984). Derived from the Greek opsismos ("late"), the name "opsismodysplasia" describes a delay in bone maturation . In addition to this delay, the disorder is characterized by micromelia (short or undersized bones), particularly of the hands and feet, delay of ossification (bone cell formation), platyspondyly (flattened vertebrae ), irregular metaphyses , an array of facial aberrations and respiratory distress related to chronic infection . Opsismodysplasia is congenital , being apparent at birth. It has a variable mortality, with some affected individuals living to adulthood. The disorder is rare , with an incidence of less than 1 per 1,000,000 worldwide.
Opsismodysplasia is a rare skeletal dysplasia characterized by congenital short stature and characteristic craniofacial abnormalities. Clinical signs observed at birth include short limbs, small hands and feet, relative macrocephaly with a large anterior fontanel (the space between the front bones of the skull), and characteristic craniofacial abnormalities including a prominent brow, depressed nasal bridge, a small anteverted nose, and a relatively long philtrum . Children with opsismodysplasia are at an increased risk for respiratory infections and respiratory failure. This condition is caused by mutations in the INPPL1 the gene. It is inherited in an autosomal recessive manner.
Cailloux et al. (2000) investigated 28 SPG families without large PLP duplications or deletions by PCR amplification and sequencing of the 7 coding regions and the splice sites of the PLP gene.
A rare, X-linked leukodystrophy characterized primarily by spastic gait and autonomic dysfunction. When additional central nervous system (CNS) signs, such as intellectual deficit, ataxia, or extrapyramidal signs, are present, the syndrome is referred to as complicated SPG. Epidemiology The prevalence and incidence of SPG2 have not been reported, but as part of the Pelizaeus-Merzbacher (PMD; see this term) spectrum, SPG2 roughly accounts for about 20 % of cases. There have been approximately 20 cases published on SPG2. SPG2 affects males but some female heterozygotes presenting in adulthood with a milder phenotype have also been reported. Clinical description SPG2 spans a continuum of phenotypes that goes from pure to complicated SPG2.
Spastic paraplegia type 2 is part of a group of genetic disorders known as hereditary spastic paraplegias. These disorders are characterized by progressive muscle stiffness (spasticity) and the development of paralysis of the lower limbs (paraplegia). Hereditary spastic paraplegias are divided into two types: pure and complex. The pure types involve the lower limbs. The complex types involve the lower limbs and can also affect the upper limbs to a lesser degree; the structure or functioning of the brain; and the nerves connecting the brain and spinal cord to muscles and sensory cells that detect sensations such as touch, pain, heat, and sound (the peripheral nervous system). Spastic paraplegia type 2 can occur in either the pure or complex form.
This protein, found in the retina and the skin, exists in fractional percentages of the normal levels found in homozygous Lp/Lp horses and so compromises the basic chemical reaction for nerve impulse transmission. [7] References [ edit ] ^ " Nyctalopia Origin" . dictionary.com . Retrieved 28 September 2015 . ^ Quesnel, NM; Lovasik, JV; Ferremi, C; Boileau, M; Ieraci, C (Jun 2004).
Ramaswami et al. (1998) screened 65 children with hypochondroplasia diagnosed by clinical and radiologic criteria for 2 previously described mutations, 1620C-A (134934.0010) and 1620C-G (134934.0012), in FGFR3; 28 (43%) of the 65 patients were heterozygous for the 1620C-A transversion, resulting in a lys540-to-asn substitution in the tyrosine kinase domain of FGFR3.
Summary Clinical characteristics. Hypochondroplasia is a skeletal dysplasia characterized by short stature; stocky build; disproportionately short arms and legs; broad, short hands and feet; mild joint laxity; and macrocephaly. Radiologic features include shortening of long bones with mild metaphyseal flare; narrowing of the inferior lumbar interpedicular distances; short, broad femoral neck; and squared, shortened ilia. The skeletal features are very similar to those seen in achondroplasia but tend to be milder. Medical complications common to achondroplasia (e.g., spinal stenosis, tibial bowing, obstructive apnea) occur less frequently in hypochondroplasia but intellectual disability and epilepsy may be more prevalent. Children usually present as toddlers or at early school age with decreased growth velocity leading to short stature and limb disproportion.
A primary bone dysplasia with micromelia characterized by disproportionate short stature, mild lumbar lordosis and limited extension of the elbow joints. Epidemiology Hypochondroplasia estimated incidence is 1/50,000. The exact prevalence is unknown. Clinical description Hypochondroplasia is a skeletal dysplasia characterized by short stature; stocky build; disproportionately short arms and legs; broad, short hands and feet; mild joint laxity; and macrocephaly. Radiologic features include shortening of long bones with mild metaphyseal flare; narrowing of the inferior lumbar interpedicular distances; short, broad femoral neck; and squared, shortened ilia. The skeletal features are very similar to those seen in achondroplasia but tend to be milder.
Hypochondroplasia Hypochondroplasia is autosomal dominant in inheritance. Specialty Medical genetics Symptoms Skeletal dysplasia [1] Causes FGFR3 gene mutation [2] Diagnostic method Physical finding, X-ray [3] Treatment Special education, Laminectomy [1] Hypochondroplasia ( HCH ) is a developmental disorder caused by an autosomal dominant genetic defect in the fibroblast growth factor receptor 3 gene ( FGFR3 ) that results in a disproportionately short stature, micromelia [3] and a head that appears large in comparison with the underdeveloped portions of the body. It is classified as short-limbed dwarfism . [2] [4] Contents 1 Signs and symptoms 2 Cause 3 Pathophysiology 4 Diagnosis 5 Treatment 6 Prognosis 7 See also 8 References 9 Further reading 10 External links Signs and symptoms [ edit ] Individuals affected by this disorder appear normal at birth . As the infant grows, however, their arms and legs do not develop properly, and their body becomes thicker and shorter than normal. [3] The following are characteristics consistent with this condition: [1] Brachydactyly Short stature Micromelia Skeletal dysplasia Abnormality of femur Cause [ edit ] Hypochondroplasia is transmitted as an autosomal dominant trait affecting the FGFR3 gene on chromosome 4p 16.3. There is currently no cure for this condition. [2] Pathophysiology [ edit ] TYK This disorder results from mutations in the proximal tyrosine kinase domain of the FGFR3 gene. [3] This gene plays an important role in embryonic development , playing a part in regulating activities such as cell division , migration and differentiation . [ medical citation needed ] Hypochondroplasia can result from p.
Hypochondroplasia is a form of skeletal disease characterized by very short stature. Hypochondroplasia is similar to achondroplasia , but the features tend to be milder. People with hypochondroplasia usually have very short stature, large head, accentuated lordosis, short arms and legs, and broad, short hands and feet. Other features include a limited range of motion in the elbows, lordosis , and bowed legs. Uncommon symptoms may include learning difficulties and convulsions. Hypochondroplasia is caused by mutations in the FGFR3 gene and is inherited in an autosomal dominant fashion.
Hypochondroplasia is a form of short-limbed dwarfism. This condition affects the conversion of cartilage into bone (a process called ossification), particularly in the long bones of the arms and legs. Hypochondroplasia is similar to another skeletal disorder called achondroplasia, but the features tend to be milder. All people with hypochondroplasia have short stature. The adult height for men with this condition ranges from 138 centimeters to 165 centimeters (4 feet, 6 inches to 5 feet, 5 inches). The height range for adult women is 128 centimeters to 151 centimeters (4 feet, 2 inches to 4 feet, 11 inches). People with hypochondroplasia have short arms and legs and broad, short hands and feet .