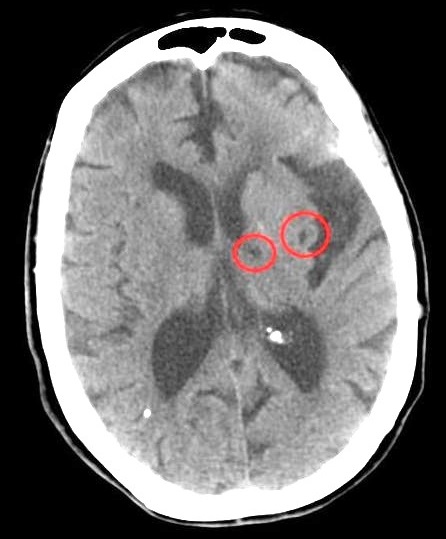
Patients who present with symptoms of a lacunar stroke, but who have not yet had diagnostic imaging performed, may be described as suffering from lacunar stroke syndrome ( LACS ). Much of the current knowledge of lacunar strokes comes from C. ... He observed "lacunae" (empty spaces) in the deep brain structures after occlusion of 200–800 μm penetrating arteries and connected them with five classic syndromes . These syndromes are still noted today, though lacunar infarcts are diagnosed based on clinical judgment and radiologic imaging. ... The classic syndromes are as follows: Name Location of infarct Presentation Pure motor stroke/hemiparesis (most common lacunar syndrome: 33–50%) posterior limb of the internal capsule , basilar part of pons , corona radiata It is marked by hemiparesis or hemiplegia that typically affects the face, arm, or leg of the side of the body opposite the location of the infarct. ... Mixed sensorimotor stroke thalamus and adjacent posterior internal capsule , lateral pons This lacunar syndrome involves hemiparesis or hemiplegia (weakness) with sensory impairment in the contralateral side. ... External links [ edit ] Classification D ICD - 10 : G46.5 - G46.7 ICD - 9-CM : 434.91 MeSH : D020520 DiseasesDB : 31186 External resources eMedicine : pmr/63 v t e Cerebrovascular diseases including stroke Ischaemic stroke Brain Anterior cerebral artery syndrome Middle cerebral artery syndrome Posterior cerebral artery syndrome Amaurosis fugax Moyamoya disease Dejerine–Roussy syndrome Watershed stroke Lacunar stroke Brain stem Brainstem stroke syndrome Medulla Medial medullary syndrome Lateral medullary syndrome Pons Medial pontine syndrome / Foville's Lateral pontine syndrome / Millard-Gubler Midbrain Weber's syndrome Benedikt syndrome Claude's syndrome Cerebellum Cerebellar stroke syndrome Extracranial arteries Carotid artery stenosis precerebral Anterior spinal artery syndrome Vertebrobasilar insufficiency Subclavian steal syndrome Classification Brain ischemia Cerebral infarction Classification Transient ischemic attack Total anterior circulation infarct Partial anterior circulation infarct Other CADASIL Binswanger's disease Transient global amnesia Haemorrhagic stroke Extra-axial Epidural Subdural Subarachnoid Cerebral/Intra-axial Intraventricular Brainstem Duret haemorrhages General Intracranial hemorrhage Aneurysm Intracranial aneurysm Charcot–Bouchard aneurysm Other Cerebral vasculitis Cerebral venous sinus thrombosis
"Empty nose syndrome". Curr Allergy Asthma Rep . 15 : 493. doi : 10.1007/s11882-014-0493-x . ... CS1 maint: multiple names: authors list ( link ) ^ a b Payne SC (2009). "Empty nose syndrome: what are we really talking about?". ... "Investigating hyperventilation syndrome in patients suffering from empty nose syndrome". ... PMID 29164622 . ^ Velasquez N, Thamboo A, Habib A-RR, Huang Z, Nayak JV (2017). "The Empty Nose Syndrome 6‐item Questionnaire: a validated 6‐item questionnaire as a diagnostic aid for empty nose syndrome patients". ... April 14, 2016 Is Empty Nose Syndrome Real? And If Not, Why Are People Killing Themselves Over It External links [ edit ] Classification D ICD - 10 : Xxx.x ICD - 9-CM : xxx American Rhinologic Society: Empty nose syndrome
Clinical Features Christian et al. (1975) described a mother and daughter with a syndrome of metacarpal and metatarsal asymmetry, platyspondyly, carpal and tarsal fusions, syndactyly, articular dysplasia, and platyspondyly. ... Garcia-Cruz et al. (1995) reported a mother and daughter with a similar syndrome characterized by proximal and distal flexion contractures in the phalanges and by brachydactyly, clinodactyly, and ulnar and radial subdislocations of the fingers. ... The authors referred to the disorder as Christian spondylodigital syndrome. Skel - Metacarpal and metatarsal asymmetry - Carpal and tarsal fusions - Platyspondyly Inheritance - Autosomal recessive ▲ Close
Ehlers-Danlos syndromes (EDS) form a heterogeneous group of hereditary connective tissue diseases characterized by joint hyperlaxity, cutaneous hyperelasticity and tissue fragility. ... Most patients have short stature and orofacial characteristics such as micrognathia, gingival hyperplasia with varying degrees of hyperkeratosis, and agenesis or microdontia of multiple teeth, accompanied sometimes by increased sensitivity to infection. Etiology The syndrome appears to be genetically heterogeneous. However, analysis of several patients has led to the identification of a potential gene locus on chromosome 12p13. Genetic counseling The syndrome is transmitted in an autosomal dominant manner.
Genetic Heterogeneity of Ehlers-Danlos Syndrome, Periodontal Type Ehlers-Danlos syndrome periodontal type 2 (EDSPD2; 617174) is caused by mutation in the C1S gene (120580) on chromosome 12p13. ... Nomenclature Beighton et al. (1998) reported on a revised nosology of the Ehlers-Danlos syndromes, designated the Villefranche classification. ... The appearance resembled that in the Ehlers-Danlos syndrome. The knees showed small 'cigarette-paper scars.' ... Stewart et al. (1977) described a case of this syndrome. The father and a half brother were also affected. ... Indeed, the father had been thought to have Werner syndrome (277700). He had become edentulous at age 16 years because of severe periodontal disease.
A number sign (#) is used with this entry because of evidence that Ehlers-Danlos syndrome periodontal type 2 (EDSPD2) is caused by heterozygous mutation in the C1S gene (120580) on chromosome 12p13. ... Clinical Features Kapferer-Seebacher et al. (2016) studied 2 families with Ehlers-Danlos syndrome and periodontitis. In the first family (family 16), the 45-year-old female proband exhibited mild elastic skin, easy bruising, fragile skin with pretibial discoloration, and early-onset periodontitis resulting in tooth loss in her teens.
One family was black, the other white. See Fraser syndrome (219000) for a comparable, although seemingly distinct, syndrome of malformations in sibs. It now seems clear that the first of the families reported by Bowen et al. (1964), that contributed by Zellweger, had cerebrohepatorenal syndrome (see 214100). The nature of the defect in the second family is not certain.
(December 2014). "Exploding head syndrome". Sleep Medicine Reviews . 18 (6): 489–493. doi : 10.1016/j.smrv.2014.03.001 . ... PMID 25726283 . ^ a b c Frese, A.; Summ, O.; Evers, S. (6 June 2014). "Exploding head syndrome: Six new cases and review of the literature". ... "Characteristic symptoms and associated features of exploding head syndrome in undergraduates". Cephalalgia . 38 (3): 595–599. doi : 10.1177/0333102417702128 . ... "Did René Descartes have Exploding Head Syndrome?" . J. Clin. Sleep Med . 14 (4): 675–8. doi : 10.5664/jcsm.7068 . ... Arlington, VA: American Psychiatric Association; 2013. ^ a b c Sharpless BA (2015). "Exploding head syndrome is common in college students".
Waterhouse–Friderichsen syndrome Other names Hemorrhagic adrenalitis The adrenal glands lie above the kidneys Specialty Endocrinology Causes Bacterial infection Waterhouse–Friderichsen syndrome ( WFS ) is defined as adrenal gland failure due to bleeding into the adrenal glands, commonly caused by severe bacterial infection. ... Amputations, reconstructive surgery, and tissue grafting are sometimes needed as a result of tissue necrosis (typically of the extremities) caused by the infection. [ citation needed ] History [ edit ] Waterhouse–Friderichsen syndrome is named after Rupert Waterhouse (1873–1958), an English physician , and Carl Friderichsen (1886–1979), a Danish pediatrician , who wrote papers on the syndrome, which had been previously described. [9] [10] References [ edit ] ^ "Waterhouse–Friderichsen syndrome" . ... ISBN 978-0-7216-0187-8 . ^ "Waterhouse-Friderichsen syndrome" . MedlinePlus Medical Encyclopedia . ... "Staphylococcus aureus sepsis and the Waterhouse-Friderichsen syndrome in children". N Engl J Med . 353 (12): 1245–51. doi : 10.1056/NEJMoa044194 . ... PMID 3873065 . ^ McKinney WP, Agner RC (December 1989). "Waterhouse-Friderichsen syndrome caused by Haemophilus influenzae type b in an immunocompetent young adult".
Congenital heart disease characterized by underdevelopment of the structures on the right side of the heart commonly associated with atrial septal defect Hypoplastic right heart syndrome Specialty Cardiology Hypoplastic right heart syndrome is a congenital heart defect in which the right atrium and right ventricle are underdeveloped. ... It can be associated with aortic stenosis . [10] References [ edit ] ^ "Hypoplastic right heart syndrome" . Genetic and Rare Diseases Information Center (GARD) . Retrieved 17 April 2018 . ^ a b c CHD-UK, Hypoplastic Right heart Syndrome (HRHS) , 2007-2015. 25 April 2015. ^ Mayo Foundation for Medical Education and Research, Hypoplastic Left Heart Syndrome , 1998-2015. 12 April 2015. ^ Heart Contraction and Blood Flow Archived 2014-10-07 at the Wayback Machine . National Institutes of Health . ^ a b "Hypoplastic Right Heart Syndrome (HRHS): Diagnosis & Treatment | SSM Health" . ^ Barrett, Heidi (January 11, 2016). ... "Aortic stenosis in hypoplastic right heart syndrome, associated with interstitial deletion of chromosome 2".
Hypoplastic right-heart syndrome (HRHS) is a rare, cyanotic congenital heart malformation (see this term) caused by underdevelopment of the right-sided heart structures (tricuspid valve, RV, pulmonary valve, and pulmonary artery) commonly associated with an atrial septal defect, ostium secundum type (see this term).
Loin pain hematuria syndrome Sagittal section of the kidney and its capsule. ... In some cases, loin pain-haematuria syndrome occurs after a bladder infection with involvement of the kidney. ... Treatment of loin pain-hematuria syndrome (LPHS) typically consists of pain management. ... PMID 8770964 . ^ a b c Hebert, LA. Loin pain-hematuria syndrome. In: Forman, JP (Ed). UpToDate. ... "Renal autotransplantation for the loin pain-hematuria syndrome: long-term followup of 26 cases".
Clinical Features Sugio and Kajii (1984) described a kindred in which 9 persons in 4 generations showed a syndrome of sparse hair, beaked nose, long upper lip, and severe metacarpophalangeal shortening. They reported the condition as an example of Ruvalcaba syndrome (180870); however, as pointed out by Hunter (1985), the disorder could be differentiated from Ruvalcaba syndrome by the absence of mental retardation and microcephaly and the presence of other changes resembling those of trichorhinophalangeal syndrome I (190350). ... Niikawa and Kamei (1986) reported on a sporadic case who had similar manifestations and proposed that the condition should be recognized as a new syndrome known as TRPS type III, or Sugio-Kajii syndrome. ... INHERITANCE - Autosomal dominant GROWTH Height - Short stature - Normal birth length Weight - Normal birth weight HEAD & NECK Face - Long, flat philtrum Ears - Protruding ears Eyes - Laterally sparse eyebrows Nose - Pear-shaped nose - Hypoplastic alae nasi Mouth - Thin upper lip Teeth - Crowded teeth - Supernumerary teeth SKELETAL - Mild osteopenia - Delayed bone age before puberty - Accelerated bone age after puberty Spine - Scoliosis Pelvis - Coxa plana - Coxa magna Limbs - Absence of exostoses Hands - Short hands - Cone-shaped epiphyses (middle phalanges) - Severe brachydactyly - Short metacarpals - Short phalanges Feet - Short feet - Short metatarsals SKIN, NAILS, & HAIR Hair - Sparse hair - Laterally sparse eyebrow NEUROLOGIC Central Nervous System - Normal intelligence MISCELLANEOUS - Cone-shaped epiphyses usually not present before age 2 years - Allelic to TRP1 ( 190350 ) - TRP2 (Langer-Giedion syndrome, 150230 ) is a microdeletion syndrome involving deletions of both the TRPS1 ( 604386 ) and EXT1 ( 608177 ) genes MOLECULAR BASIS - Caused by mutations in the zinc finger transcription factor TRPS1 gene (TRPS1, 604386.0007 ) ▲ Close
Anophthalmia-megalocornea-cardiopathy-skeletal anomalies syndrome is a multiple congenital anomalies syndrome, reported in the offsprings of a consanguineous couple and characterized by multiple congenital skeletal (dolichocephaly, skull asymmetry, camptodactyly, clubfoot), muscular (muscle hypoplasia), ocular (anophthalmia, buphthalmos, retinal detachment, aniridia (see this term)) and cardiac (prolapse of tricuspid valves, mitral and tricuspid insufficiency) abnormalities.
Chromosome 19q13.11 deletion syndrome is a chromosome abnormality that occurs when there is a missing ( deleted ) copy of genetic material on chromosome 19 at a location designated q13.11. ... To date, all cases of chromosome 19q13.11 deletion syndrome appear to be sporadic and diagnosed in people with no family history of the condition.
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome (chr19:39,803,651-40,127,916, NCBI36). See also proximal chromosome 19q13.11 deletion syndrome (617219), which shows some phenotypic overlap. ... Using array comparative genomic hybridization (CGH), Malan et al. (2009) identified 3 unrelated patients with syndromic mental retardation associated with interstitial microdeletions involving chromosome 19q13.11. ... Chowdhury et al. (2014) reported 2 unrelated patients, a girl and a boy (patients 1 and 2), with 19q13.11 deletion syndrome. The patients had low birth weight, feeding difficulties with poor postnatal growth, and delayed development with speech delay. ... By array CGH analysis of 3 unrelated individuals with syndromic mental retardation, Malan et al. (2009) identified interstitial overlapping deletions of chromosome 19q12-q13.12, 19q13.11-q13.12, and 19q12-q13.11, respectively.
The 19q13.11 microdeletion is characterized by several major features including pre and postnatal growth retardation, slender habitus, severe postnatal feeding difficulties, microcephaly, intellectual deficit with speech disturbance, hypospadias and ectodermal dysplasia presented by scalp aplasia, thin and sparse hair, eyebrows and eyelashes, thin and dry skin and dysplasic nails. Epidemiology To date, the syndrome has been identified in five patients. ... Haploinsufficiency of one or more genes in the 19q13.11 region could cause this microdeletion syndrome. The careful clinical examination and the molecular characterization of additional patients with a similar chromosomal anomaly are needed to further delineate the clinical features and refine the minimal critical region.
Microphthalmia-retinitis pigmentosa-foveoschisis-optic disc drusen syndrome is a rare, genetic, non-syndromic developmental defect of the eye disorder characterized by the association of posterior microphthalmia, retinal dystrophy compatible with retinitis pigmentosa, localized foveal schisis and optic disc drusen.
The patient had axial lengths of 15.65 mm and extreme hyperopia (+15.75 diopters) as well as a thick papillomacular fold, but exhibited no night blindness or clinical signs of retinal degeneration, developmental ocular malformations, or syndromic disease. The authors observed corneal steepening that was proportional to the degree of axial foreshortening in this and other patients with posterior microphthalmia, including the patients previously studied by Aldahmesh et al. (2011).
Pelvic dysplasia-arthrogryposis of lower limbs syndrome is a rare, genetic, dysostosis syndrome characterized by intrauterine growth restriction, short stature (with short lower segment), lower limb joint contractures and muscular hypotrophy, narrow, small pelvis, lumbar hyperlordosis with scoliosis, and foot deformity (short, overlapping toes).
Clinical Features Sarralde et al. (1998) described 4 brothers and a sister from a sibship of 9 who had a seemingly distinctive syndrome with prenatal growth retardation, pelvic hypoplasia, and arthrogrypotic changes in the lower limbs.
Primary tethered cord syndrome is a genetic, non-syndromic congenital malformation of the neurenteric canal, spinal cord and column characterized by progressive neurologic deterioration (pain, sensorimotor deficits, abnormal gait, decreased tone or abnormal reflexes), musculoskeletal changes (foot deformities and asymmetry, muscle atrophy, limb weakness and numbness, gait disturbances, scoliosis) and/or genitourinary manifestations (bladder and bowel dysfunction).
Ring chromosome 12 syndrome is a rare chromosomal anomaly syndrome with a highly variable phenotype principally characterized by postnatal growth retardation, variable degrees of developmental delay and intellectual disability, microcephaly and facial dysmorphism (incl. epicanthal folds, low-set, cupped ears, prominent nose with flat nasal bridge, high arched palate, micrognathia).
Frontonasal dysplasia-severe microphthalmia-severe facial clefting syndrome is a rare, genetic, orofacial clefting malformation syndrome characterized by severe frontonasal dysplasia with complete cleft palate, facial cleft, extreme microphtalmia and hypertelorism, frequently associated with eyelid colobomata, sparse or absent eyelashes/eyebrows, wide nasal bridge with hypoplastic alae nasi, low-set, posteriorly rotated ears and caudal appendage in the sacral region.
Uz et al. (2010) stated that the phenotype in these patients was borderline between frontofacionasal dysplasia (229400) and Fryns microphthalmia syndrome (600776). Mapping Uz et al. (2010) performed genomewide homozygosity mapping of 3 affected sibs with severe frontonasal dysplasia and their unaffected consanguineous parents and identified a single large homozygous stretch spanning approximately 27 Mb on chromosome 12q21.3.
Multiple sclerosis-ichthyosis-factor VIII deficiency syndrome is characterized by the association of multiple sclerosis with lamellar ichthyosis (see this term) and hematological anomalies (beta thalassemia minor and a quantitative deficit of factor VIII-von Willebrand complex). Other clinical manifestations may include eye involvement (optic atrophy, diplopia), neuromuscular involvement (ataxia, pyramidal syndrome, gait disturbance) and sensory disorder.
Patent ductus arteriosus - bicuspid aortic valve - hand anomalies syndrome is a very rare heart-hand syndrome (see this term) that is characterized by a variety of cardiovascular anomalies including patent arterial duct, bicuspid aortic valve and pseudocoarctation of the aorta in conjunction with hand anomalies such as brachydactyly and ulnar ray derivative i.e. fifth metacarpal hypoplasia.
Given the similarity between this syndrome and Char syndrome (169100), the authors performed linkage analysis using DNA markers spanning the Char syndrome critical region at 6p21.1-p12. This analysis excluded the possibility that the family was inheriting an allelic variant of Char syndrome. Gelb et al. (1999) concluded that this is a novel heart-hand syndrome.