-
16p13.3 Microduplication Syndrome
Orphanet
16p13.3 microduplication syndrome is a rare chromosomal anomaly syndrome resulting from a partial duplication of the short arm of chromosome 16 and manifesting with a variable phenotype which is mostly characterized by: mild to moderate intellectual deficit and developmental delay (particularly speech), normal growth, short, proximally implanted thumbs and other hand and feet malformations (such as camptodactyly, syndactyly, club feet), mild arthrogryposis and characteristic facies (upslanting, narrow palpebral fissures, hypertelorism, mid face hypoplasia, bulbous nasal tip and low set ears).
-
Progressive Microcephaly-Seizures-Cortical Blindness-Developmental Delay Syndrome
Orphanet
Progressive microcephaly-seizures-cortical blindness-developmental delay syndrome is a rare, genetic, neuro-ophthalmological syndrome characterized by post-natal, progressive microcephaly and early-onset seizures, associated with delayed global development, bilateral cortical visual impairment and moderate to severe intellectual disability.
-
Brachydactyly-Long Thumb Syndrome
Orphanet
Brachydactyly - long thumb syndrome is a very rare autosomal dominant heart-hand syndrome (see this term) that is characterized by bisymmetric brachydactyly accompanied by long thumbs, joint anomalies (restriction of motion at the shoulder and metacarpophalangeal joints) and cardiac conduction defects.
-
Blepharophimosis-Intellectual Disability Syndrome, Verloes Type
Orphanet
Blepharophimosis-intellectual disability syndrome, Verloes type is a rare, genetic multiple congenital anomalies/dysmorphic syndrome characterized by congenital microcephaly, severe epilepsy with hypsarrhythmia, adducted thumbs, abnormal genitalia, and normal thyroid function.
-
Tibial Hemimelia-Polysyndactyly-Triphalangeal Thumb Syndrome
Orphanet
Tibial hemimelia-polysyndactyly-triphalangeal thumb syndrome is a rare, genetic dysostosis syndrome, with marked inter- and intra-familial variation, typically characterized by triphalangeal thumbs, hand and/or foot polysyndactyly and/or absent/hypoplastic tibiae (associated with duplication of fibulae in some cases), although isolated triphalangeal thumbs have also been reported.
-
Facial Dysmorphism-Lens Dislocation-Anterior Segment Abnormalities-Spontaneous Filtering Blebs Syndrome
Orphanet
Facial dysmorphism-lens dislocation-anterior segment abnormalities-spontaneous filtering blebs syndrome is a syndromic developmental defect of the eye characterized by dislocated or subluxated crystalline lenses, anterior segment abnormalities, and distinctive facial features such as flat cheeks and a prominent, beaked nose.
-
Anhidrotic Ectodermal Dysplasia-Immunodeficiency-Osteopetrosis-Lymphedema Syndrome
Orphanet
This syndrome is characterized by severe immunodeficiency, osteopetrosis, lymphedema and anhidrotic ectodermal dysplasia. ... Clinical description The first two reported children died before three years of age from multiple infections with Gram-positive cocci, Gram-negative bacilli, mycobacteria, and fungi. Etiology The syndrome is classified as a X-linked osteopetrosis and is caused by mutations in the IKBKG ( NEMO ) gene (Xq28).
-
Fitz-Hugh-Curtis Syndrome
Gard
Fitz-Hugh-Curtis syndrome (FHCS) is a condition in which a woman has swelling of the tissue covering the liver as a result of having pelvic inflammatory disease (PID). ... FHCS is usually caused by an infection of chlamydia or gonorrhea that leads to PID; it is not known why PID progresses to FHCS in some women. Fitz-Hugh-Curtis syndrome is treated with antibiotics.

-
12p12.1 Microdeletion Syndrome
Orphanet
12p12.1 microdeletion syndrome is a rare chromosomal anomaly syndrome, resulting from the partial deletion of the short arm of chromosome 12, characterized by intellectual disability, global developmental delay with prominent language impairment, behavioral abnormalities and mild facial dysmorphism (incl. frontal bossing, downslanting palpebral fissures, epicanthal folds, broad, depressed nasal bridge with bulbous nasal tip, low-set ears with underdeveloped helices).

-
Coloboma Of Macula-Brachydactyly Type B Syndrome
Orphanet
Coloboma of macula - brachydactyly type B or Sorsby syndrome is a malformation syndrome characterized by the combination of bilateral coloboma of macula with horizontal pendular nystagmus and severe visual loss, and brachydactyly type B (see these terms).
-
Split Hand Syndrome
Wikipedia
This article is about the neurological syndrome. For the congenital disorder, see split hand . Split hand syndrome Specialty Neurological In medicine , split hand syndrome is a neurological syndrome in which the hand muscles on the side of the thumb ( lateral , thenar eminence ) appear wasted , whereas the muscles on the side of the little finger ( medial , hypothenar eminence ) are spared. ... S2CID 18500530 . ^ a b Split hand syndrome . Stedman's Medical Dictionary . ... Can J Neurol Sci . 21 (S2): S9. ^ Wilbourn AJ (January 2000). "The "split hand syndrome " " . Muscle Nerve . 23 (1): 138. doi : 10.1002/(SICI)1097-4598(200001)23:1<138::AID-MUS22>3.0.CO;2-7 . ... Archived from the original on 2013-01-05. v t e Symptoms and signs relating to movement and gait Gait Gait abnormality CNS Scissor gait Cerebellar ataxia Festinating gait Marche à petit pas Propulsive gait Stomping gait Spastic gait Magnetic gait Truncal ataxia Muscular Myopathic gait Trendelenburg gait Pigeon gait Steppage gait Antalgic gait Coordination Ataxia Cerebellar ataxia Dysmetria Dysdiadochokinesia Pronator drift Dyssynergia Sensory ataxia Asterixis Abnormal movement Athetosis Tremor Fasciculation Fibrillation Posturing Abnormal posturing Opisthotonus Spasm Trismus Cramp Tetany Myokymia Joint locking Paralysis Flaccid paralysis Spastic paraplegia Spastic diplegia Spastic paraplegia Syndromes Monoplegia Diplegia / Paraplegia Hemiplegia Triplegia Tetraplegia / Quadruplegia General causes Upper motor neuron lesion Lower motor neuron lesion Weakness Hemiparesis Other Rachitic rosary Hyperreflexia Clasp-knife response
-
Vexas Syndrome
Wikipedia
Auto-inflammatory syndrome The VEXAS syndrome is an adult-onset autoinflammatory disease affecting males, caused by a mutation in the UBA1 gene. [1] [2] [3] [4] [5] The name derives from Vacuoles , E1 enzyme , X-linked , Autoinflammatory , Somatic . [6] The syndrome was first reported in a paper in The New England Journal of Medicine in October 2020, by Beck et al, who write: "Using a genotype -driven approach, we identified a disorder that connects seemingly unrelated adult-onset inflammatory syndromes." [7] An editorial in the same issue describes the work as a "fascinating discovery" which "is of immediate importance to rheumatologists and has far-reaching consequences of general clinical interest. It builds on previous findings suggesting that postzygotic somatic mutation may be a more frequent cause of human disease than previously recognized". [8] The podcast The Naked Scientists featured an interview with Dan Kastner about VEXAS, in an episode titled "Mink, Ivory, & a Disease Discovered Backwards" broadcast on 13 November 2020. [9] References [ edit ] ^ Onuora, Sarah (1 December 2020). "Somatic mutations cause VEXAS syndrome". Nature Reviews Rheumatology . doi : 10.1038/s41584-020-00559-x . ^ Mathews, Stephanie (4 November 2020). ... Retrieved 17 November 2020 . ^ Edwards, Erika (27 October 2020). " ' The VEXAS syndrome': Scientists discover a rare and deadly inflammatory disorder in men" . ... "Scientists use clues in the human genome to discover new inflammatory syndrome" . Genome.gov . National Human Genome Research Institute . ... External links [ edit ] Online Mendelian Inheritance in Man (OMIM): VEXAS syndrome; VEXAS - 301054 Classification D OMIM : 301054 This article about a disease , disorder, or medical condition is a stub .
-
Nephronophthisis
Orphanet
Extrarenal phenotypes are distinct but they overlap in some syndromic forms of NPHP (such as Joubert syndrome, Senior-Loken syndrome, Meckel-Gruber syndrome). ... Mutations in NPHP3 (3q22.1), NPHP4 (1p36.31), NEK8 (17q11.2) genes give rise to late-onset nephronophthisis, however these genes are associated also with the Senior-Loken syndrome and Meckel-Gruber syndrome and predispose to multiorgan polycystic disease. ... Furthermore, juvenile NPHP forms part of a spectrum of NPHP-related ciliopathies which includes Joubert syndrome, Senior-Loken syndrome, Meckel-Gruber syndrome, Bardet-Biedl syndrome and Skeletal Ciliopathies (Oral-facial-digital syndrome, Cranioectodermal dysplasia, Short-rib thoracic dysplasia).NPHP3, INVS, NPHP1, NPHP4, GLIS2, NEK8, WDR19, ANKS6, TMEM67, TTC21B, XPNPEP3, AHI1, SLC41A1, ATXN10, CNTRL, WWTR1, TMEM218, CEP290, RPGRIP1L, IQCB1, DCDC2, TRAF3IP1, ZNF423, MAPKBP1, CEP164, SDCCAG8, IFT140, NPHP3-ACAD11, PKHD1, CC2D2A, DYNC2LI1, NPHP3-AS1, FAM186B, DYNC2H1, WDR34, TTC21B-AS1, IFT43, IFT80, TMEM216, WDR60, CEP120, INTU, PAM16, RBM48, CEP83, IFT172, PRPF40B, UMOD, FAN1, INCENP, PIAS1, MUC1, NXPH1, ANPEP, BCL2, MKS1, ACTN4, MKKS, MALL, CYS1, DAP, SLC22A12, ANKS3, SCLT1, PACRG, CTNNB1, GLIS3, COL4A1, TIMM8A, RCC1, CDK5, AATF, AVPR2, AGXT, LINC01672, ALDH3A2, ACE, FN1, WT1, IL1A, VIM, MLRL, TNF, ADAMTS9, RPGRIP1, TIMP2, HNF1B, HNF1A, SLC4A1, PAX2, TINAG, MLYCD, TCTN2, KIF3A, IL1B, CSPP1

-
Parkinson-Dementia Syndrome
Omim
Clinical Features Mata et al. (1983) described 2 brothers and a sister with a 'new' Parkinson-dementia syndrome. The disorder, characterized also by ophthalmoparesis and pyramidal signs, came on in the third decade and progressed for several years. ... Ohara et al. (1994) described 2 of 5 sibs of first-cousin parents of Japanese descent who developed vertical ophthalmoparesis, dementia, a parkinsonian syndrome, jaw tremor, and bradykinesia. Both sibs had a poor response to levodopa. The authors referred to this syndrome as a progressive supranuclear palsy-like syndrome. Progressive supranuclear palsy (PSP; 601104), also known as Steele-Richardson-Olszewski syndrome, is a sporadic disorder of adult onset with supranuclear palsy of vertical gaze, followed by proximal rigidity and dementia. ... Also unlike patients with classic Steele-Richardson-Olszewski syndrome, the affected sibs stooped forward rather than having their head in an opisthotonic position, a distinctive feature in the sporadic disorder.MAPT, STX6, MOBP, EIF2AK3, SRSF2, TRA2B, SLCO1A2, TRIM11, SP1, PSPH, REG1A, SNCA, RIDA, STXBP3, MSMB, PSPN, TPO, CD8B, ASAP1, RUNX2, BPIFA2, PIK3C2G, IRF4, APOE, SLC6A3, TARDBP, LRRK2, NEFL, SOD1, CIT, C9orf72, CSF2, MAOB, GRN, LAMC2, DCTN1, STH, SMUG1, PRKN, UBB, PYCARD, NPC1, APP, TYMS, CRHR1, TH, TGM2, SLC25A38, ATXN2, GFAP, IGLON5, CST3, NPEPPS, VEGFA, RAB35, YWHAE, OGA, CXCR4, PICALM, NPC2, SNCAIP, BSN, MAP3K14, OPN1MW3, DUSP10, ARL17B, ROCK2, SCRN1, MAP4K4, NF1P1, UNC13A, DNAJB1P1, FLAD1, UBASH3B, SPECC1, FOXP2, RMDN2, ASXL1, MCIDAS, SETX, MIR132, MIR518E, GGTLC5P, GGTLC3, GGT2, OPN1MW2, CTNNBL1, SYBU, PSPC1, RMDN3, LRRC37A4P, TET2, TMEM106B, TREM2, LCMT1, PPME1, RMDN1, GGTLC4P, PSAT1, TBK1, CSDC2, LMOD1, SF3B1, MINK1, NAT1, TPI1, OPN1MW, FMR1, MTOR, FUS, GABPA, GABRG2, GBA, GGT1, EGFR, GLDC, GSTM1, NRG1, HSPA4, DNAJB1, IFNG, ERBB4, DLX1, IGFALS, CASP3, AP2A2, ANXA6, KLK3, BDNF, BNIP1, BRCA1, CBS, ACE, CDK5, CHI3L1, CLU, CRP, CTSS, CYP2D6, IGF1, IL2, TP53BP1, MAP2K4, PSEN2, PTEN, PTPRC, RAPSN, ROCK1, ATXN8OS, NAT2, PROS1, SPOCK1, SPP1, TCOF1, TGFB1, TGM1, TNF, PSEN1, PRNP, IL6, NR4A2, IRS1, MUSK, NFE2L2, NGF, NOS1, NSF, PAEP, PTPA, PAFAH1B1, PDK1, PIN1, PLAG1, PLCG2, PLXNA2, ATXN2-AS
-
Auriculocondylar Syndrome 3
Omim
A number sign (#) is used with this entry because of evidence that auriculocondylar syndrome-3 (ARCND3) is caused by homozygous mutation in the EDN1 gene (131240) on chromosome 6p24. Heterozygous mutation in EDN1 causes isolated question mark ears (612798). Description Auriculocondylar syndrome (ARCND) is a rare craniofacial disorder involving first and second pharyngeal arch derivatives and includes the key features of micrognathia, temporomandibular joint and condyle anomalies, microstomia, prominent cheeks, and question mark ears (QMEs). ... For a general phenotypic description and discussion of genetic heterogeneity of auriculocondylar syndrome, see ARCND1 (602483). Clinical Features Guion-Almeida et al. (2002) described a 9-year-old boy with auriculocondylar syndrome whose parents were consanguineous. ... Gordon et al. (2013) studied a brother and sister ('case 10'), born to healthy first-cousin parents, who had auriculocondylar syndrome. The brother presented with bifid uvula, laryngeal cleft, short velum, retrognathia, a typical QME on the right, a severely dysmorphic left ear, and an aneurysm of the vein of Galen, while his sister displayed a left QME, over-folded helix on the right, glossoptosis, and mandibular hypoplasia requiring distraction. ... Sanger sequencing of EDN1 in a 23-year-old man with auriculocondylar syndrome who was originally described by Guion-Almeida et al. (2002) (patient 2) revealed homozygosity for a missense mutation (P77H; 131240.0003); his unaffected mother was heterozygous for the mutation.
-
Dopamine Transporter Deficiency Syndrome
Medlineplus
Dopamine transporter deficiency syndrome is a rare movement disorder. The condition is also known as infantile parkinsonism-dystonia because the problems with movement (dystonia and parkinsonism, described below) usually start in infancy and worsen over time. ... People with dopamine transporter deficiency syndrome develop a pattern of involuntary, sustained muscle contractions known as dystonia. ... People with dopamine transporter deficiency syndrome may have a shortened lifespan, although the long-term effects of this condition are not fully understood. ... Frequency Dopamine transporter deficiency syndrome appears to be a rare disease; only about 20 affected individuals have been described in the medical literature. ... Causes Dopamine transporter deficiency syndrome is caused by mutations in the SLC6A3 gene.
-
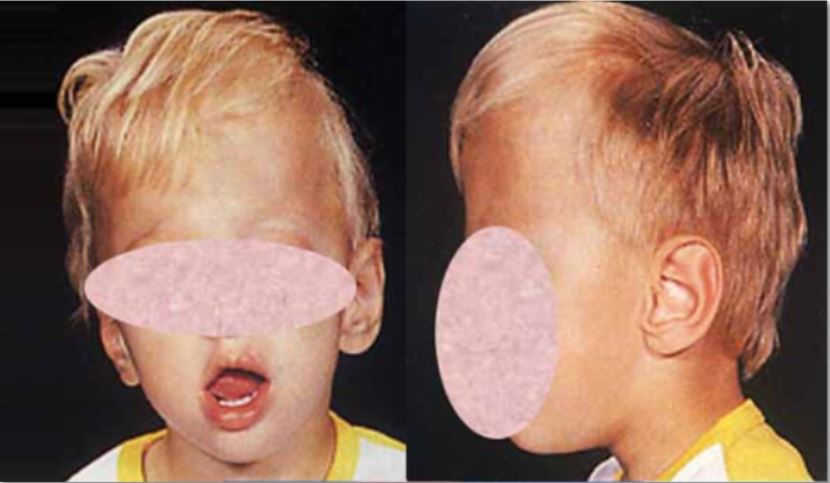
Oxycephaly
Wikipedia
Oxycephaly Other names Turricephaly , [1] Acrocephaly , Hypsicephaly , [1] Oxycephalia , [1] Steeple head , [1] Tower head , [1] Tower skull , High-head syndrome , Turmschädel , [2] Specialty Medical genetics Oxycephaly is a type of cephalic disorder where the top of the skull is pointed or conical due to premature closure of the coronal suture plus any other suture , like the lambdoid , [3] or it may be used to describe the premature fusion of all sutures. [2] It should be differentiated from Crouzon syndrome . ... External links [ edit ] Classification D ICD - 10 : Q75.0 ICD - 9-CM : 756.0 OMIM : 123100 External resources Orphanet : 63440 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum This article about a congenital malformation is a stub .ZIC1, ERF, TWIST1, FGFR2, ALX4, EFNB1, EZH2, FGFR1, WDR35, TCF12, IFT122, SMAD6, NELL1, FREM1, BBS9, BMP2, SEC24D, SLC12A6, ARVCF, SLC25A24, RAB23, SEC24C, FERMT1, TBX1, UFD1, HIRA, RREB1, PEX5, P4HB, GPX4, GP1BB, FGFR3, MEGF8, COMT, JMJD1C
-
Chromosome 18p Deletion Syndrome
Omim
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome. Clinical Features The 18p- syndrome was first described in 1963 by de Grouchy et al. ... Buhler et al. (1964) reported an early case of 18p- syndrome. In addition to the typical manifestations of that syndrome, the girl had an abnormality of thyroxine synthesis, most likely a coupling defect (Buhler, 1983). Since the proband was the youngest of 7 sibs and none of the other sibs had a thyroxine defect, Buhler (1983) suggested that the deletion may have 'uncovered' a heterozygous mutation inherited from 1 parent and that the gene locus is on the short arm of chromosome 18. Three patients with 18p- syndrome and hypopituitarism have been reported (Leisti et al., 1973; Buffoni et al., 1976; Artman et al., 1992), suggesting that a factor on chromosome 18 may in some way be involved. ... INHERITANCE - Autosomal dominant GROWTH Height - Short stature Weight - Low birth weight HEAD & NECK Face - Round face Ears - Large ears - Dysplastic ears Eyes - Hypertelorism Nose - Broad nasal bridge - Upturned nostrils Mouth - Micrognathia - High palate Teeth - Malaligned teeth Neck - Redundant neck skin GENITOURINARY External Genitalia (Male) - Micropenis - Hypoplastic testes - Gonadal dysgenesis External Genitalia (Female) - Hypoplastic genitalia Internal Genitalia (Male) - Cryptorchidism Internal Genitalia (Female) - Gonadal dysgenesis SKELETAL Hands - Clinodactyly NEUROLOGIC Central Nervous System - Developmental delay - Mental retardation - Dystonia LABORATORY ABNORMALITIES - Deletion of chromosome 18p11.2 - Contiguous gene deletion syndrome MISCELLANEOUS - Many cases due to de novo mutation MOLECULAR BASIS - Contiguous gene syndrome caused by deletion of 18p11.21-p11.1 ▲ Close

-
Multiple Mitochondrial Dysfunctions Syndrome
Medlineplus
Multiple mitochondrial dysfunctions syndrome is characterized by impairment of cellular structures called mitochondria, which are the energy-producing centers of cells. While certain mitochondrial disorders are caused by impairment of a single stage of energy production, individuals with multiple mitochondrial dysfunctions syndrome have reduced function of more than one stage. ... Some babies with multiple mitochondrial dysfunctions syndrome have high blood pressure in the blood vessels that connect to the lungs (pulmonary hypertension) or weakening of the heart muscle (cardiomyopathy). Frequency Multiple mitochondrial dysfunctions syndrome is a rare condition; its prevalence is unknown. ... Causes Multiple mitochondrial dysfunctions syndrome can be caused by mutations in the NFU1 or BOLA3 gene.
-
Mandibulofacial Dysostosis-Microcephaly Syndrome
Orphanet
A rare genetic, multiple congenital malformation syndrome characterized by malar and mandibular hypoplasia, microcephaly, ear malformations with associated conductive hearing loss, distinctive facial dysmorphism (with significantly overlap to Treacher Collins syndrome), developmental delay, and intellectual disability. ... Clinical description Mandibulofacial dysostosis-microcephaly syndrome (MFDM) has a wide range of manifestations. ... Differential diagnosis The differential diagnosis includes Treacher Collins, Nager and CHARGE syndromes and oculo-auriculo-vertebral spectrum. ... Genetic counseling should be offered to affected individuals, informing them of the 50% chance of offspring inheriting the disease-causing mutation and therefore being affected with the syndrome. The syndrome displays high penetrance but variable expressivity. ... The functional repercussions of the syndrome are highly variable with some patients being intellectually normal, some living semi-independently and employed, and some being nonverbal and in need of significant assistance. * European Reference Network




