ACTG2- related disorders are inherited in an autosomal dominant manner. While the exact proportion of inherited vs de novo pathogenic variants is unknown, current data suggest that de novo pathogenic variants are common. ... Sequence analysis of ACTG2 is performed first. Deletion/duplication analysis may be considered if no pathogenic variant is found; however, all pathogenic variants identified to date are missense variants, and no exon or whole-gene deletions/duplications have been identified as a cause of ACTG2 -related disorders. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Affected infants frequently present in the first two months of life with findings of impaired intestinal motility such as failure to pass meconium within the first 48 hours of life, constipation, emesis, abdominal pain or distention, and occasionally diarrhea. ... Inheritance can be either autosomal dominant (mutation of EDNRB , EDN3 , or SOX10 ) or autosomal recessive (mutation of EDNRB or EDN3 ).
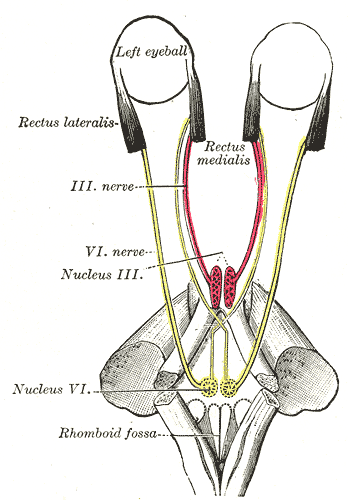
Cause [ edit ] Because the nerve emerges near the bottom of the brain , it is often the first nerve compressed when there is any rise in intracranial pressure . ... Limitations of eye movements are confined to abduction of the affected eye (or abduction of both eyes if bilateral) and the size of the resulting convergent squint or esotropia is always larger on distance fixation - where the lateral recti are more active - than on near fixation - where the medial recti are dominant. Abduction limitations that mimic VIth nerve palsy may result secondary to surgery, to trauma or as a result of other conditions such as myasthenia gravis or thyroid eye disease . ... The resultant palsy is identified through loss of lateral gaze after application of the orthosis and is the most common cranial nerve injury associated with this device. [9] Management [ edit ] The first aims of management should be to identify and treat the cause of the condition, where this is possible, and to relieve the patient's symptoms, where present. In children, who rarely appreciate diplopia , the aim will be to maintain binocular vision and, thus, promote proper visual development. [ citation needed ] Thereafter, a period of observation of around 6 months is appropriate before any further intervention, as some palsies will recover without the need for surgery. [ citation needed ] Symptom relief and/or binocular vision maintenance [ edit ] This is most commonly achieved through the use of fresnel prisms. ... Where there is complete paralysis, the preferred option is to perform vertical muscle transposition procedures such as Jensen's, Hummelheim's or whole muscle transposition, with the aim of using the functioning inferior and superior recti to gain some degree of abduction. [12] [13] [14] An alternative approach is to operate on both the lateral and medial rectii of the affected eye, with the aim of stabilising it at the midline, thus giving single vision straight ahead but potentially diplopia on both far left and right gaze.
Sixth nerve palsy is a nerve disorder that occurs when the sixth cranial nerve is damaged. The disorder prevents some of the muscles that control eye movement from working properly. Affected people cannot turn the eye outwards toward the ear. Other signs and symptoms may include double vision, headaches, and pain around the eye. Sixth nerve palsy may be caused by many things, including stroke, brain aneurysm , diabetic neuropathy , trauma, infections, inflammation, tumors, migraine headaches or intracranial pressure . Eye patches, glasses, corticosteroids, and/or botulinum toxin may be used to ease symptoms.
Andrews' Diseases of the Skin: Clinical Dermatology . (10th ed.). Saunders. Page 512. ISBN 0-7216-2921-0 . This cutaneous condition article is a stub .
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( February 2017 ) Penile torsion is a fairly common congenital condition with male infants.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( September 2014 ) Work-related musculoskeletal disorders Other names WMSDs Work-related musculoskeletal disorders are disorders of the muscles, skeleton, and related tissues.
Find sources: "Chromosome 2q deletion" – news · newspapers · books · scholar · JSTOR ( December 2018 ) ( Learn how and when to remove this template message ) This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( March 2018 ) Chromosome 2q deletion is a chromosome abnormality that occurs when there is a missing copy of the genetic material located on the long arm (q) of chromosome 2 .
Chromosome 2q deletion is a chromosome abnormality that occurs when there is a missing copy of the genetic material located on the long arm (q) of chromosome 2 . The severity of the condition and the signs and symptoms depend on the size and location of the deletion and which genes are involved. Features that often occur in people with chromosome 2q deletion include developmental delay, intellectual disability, behavioral problems, and distinctive facial features. Most cases are not inherited, but people can pass the deletion on to their children. Treatment is based on the signs and symptoms present in each person.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( January 2021 ) Pneumothorax ex vacuo Specialty Pulmonology Pneumothorax ex vacuo is a rare type of pneumothorax which forms adjacent to an atelectatic lobe . [1] It is seen preferentially with atelectasis of the right upper lobe and is the result of rapid atelectasis producing an abrupt decrease in the intrapleural pressure with subsequent release of nitrogen from pleural capillaries .
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( February 2017 ) Nasal septal abscess Nasal septum(normal) Specialty ENT surgery Nasal septal abscess is a condition of the nasal septum [1] in which there is a collection of pus between the mucoperichondrium and septal cartilage .
Treatment for chromosome 10p deletion is based on the signs and symptoms in each person. This page is meant to provide general information about 10p deletions.
The DiGeorge syndrome (DGS; 188400) and velocardiofacial syndrome (VCFS; 192430) may present many clinical problems, including cardiac defects, hypoparathyroidism, T-cell immunodeficiency, and facial dysmorphism. They are frequently associated with deletions within 22q11.2 (accounting in part for the designation CATCH22), but a number of cases have no detectable molecular defect of this region. Daw et al. (1996) stated that a number of single case reports with deletions of 10p suggested genetic heterogeneity of DGS. They compared the regions of hemizygosity in 4 patients with terminal deletions of 10p (1 patient with hypoparathyroidism and 3 with DGS) and 1 patient with VCFS and a large interstitial deletion. Fluorescence in situ hybridization (FISH) analysis demonstrated that these patients had overlapping deletions at the 10p13/10p14 boundary.
Distal monosomy 10p is a rare chromosomal disorder in which the tip of the short arm (p arm) of chromosome 10 is deleted resulting in a variable phenotype depending on the size of the deletion. The deletion may involve only the terminal 10p15 band, or extend towards the centromere to bands 10p14 or 10p13. Epidemiology Around 50 cases of pure distal monosomy 10p have been reported. Clinical description Distal monosomy 10p encompassing the 10p13 band is associated with cardiac malformations and immune anomalies that overlap with the anomalies reported in the deletion 22q11 syndrome (DiGeorge syndrome/velocardiofacial syndrome spectrum (DGS/VCFS); see this term) with hypoparathyroidism, hypocalcemia, congenital conotruncal heart defects, thymus hypoplasia leading to T-cell deficiency and intellectual deficit. More than 25 patients have been reported with del(10)(p13). In addition to the anomalies related to the DiGeorge syndrome ( e.g. conotruncal malformation with thymic hypoplasia), these patients often show an abnormally shaped skull, microcephaly, a long face, high forehead, broad nasal bridge, downslanting palpebral fissures, anteverted nares, hand and foot abnormalities, genitourinary anomalies, hearing loss and severe psychomotor retardation, resulting in a clinical picture that clearly differs from that of the classic 22q11 deletion syndrome.
A rare superficial pemphigus disease characterized clinically by well-demarcated, localized, erythematous, scaly, hyperkeratotic, crusted plaques, with frequent butterfly distribution over the malar area of the face (but also commonly involving trunk and scalp, and less frequently the extremities, with a photoexposed distribution). Histologically, granular deposits along the dermal-epidermal junction, in addition to intercellular deposition in the upper epidermis, are observed.
CCA is inherited in an autosomal dominant manner. While many individuals with CCA have an affected parent, as many as 50% may have a de novo FBN2 pathogenic variant. ... Sequence analysis of FBN2 detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis first. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications. ... Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Distal arthrogryposes is inherited in an autosomal dominant manner with the exception of ECEL1 -related distal arthrogryposis, which is inherited in an autosomal recessive manner.
Learn more about the gene associated with Congenital contractural arachnodactyly FBN2 Inheritance Pattern This condition is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Description Congenital contractural arachnodactyly is a rare, autosomal dominant connective tissue disorder characterized by contractures, arachnodactyly, scoliosis, and crumpled ears (Hecht and Beals, 1972). ... Currarino and Friedman (1986) described 2 unrelated infants with severe CCA, both of whom died in the first year of life. Cole and Hughes (1992) described an infant with presumed CCA who also had deficiency in the right lower limb. ... A mutation in a calcium-binding EGF-like motif (612570.0001) was found by the first authors and a mutation in a TGF-binding protein-like motif (612570.0002) by the second group. ... The deafness in these animals is caused by mutations in the contiguous Na-K-2Cl cotransporter gene Slc12a2 (600840) (Dixon et al., 1999). INHERITANCE - Autosomal dominant GROWTH Other - Dolichostenomelia - Marfanoid habitus HEAD & NECK Head - Scaphocephaly - Brachycephaly - Dolichocephaly Face - Micrognathia (27%) - Frontal bossing Ears - Crumpled ear (76%) - Poorly defined conchae - Prominent crura - Folded helices Eyes - Ectopia lentis - Myopia Mouth - High-arched palate (28%) Neck - Relatively short neck CARDIOVASCULAR Heart - Mitral valve prolapse - Mitral regurgitation - Atrial septal defect - Ventricular septal defect - Bicuspid aortic valve Vascular - Patent ductus arteriosus - Aortic root dilatation CHEST Ribs Sternum Clavicles & Scapulae - Pectus carinatum SKELETAL - Osteopenia Spine - Congenital kyphoscoliosis (45%) Pelvis - Hip contractures (25%) Limbs - Elbow contractures (86%) - Knee contractures (81%) - Subluxation of patella Hands - Arachnodactyly - Camptodactyly - Ulnar deviation of fingers - Adducted thumbs - Flexion contractures of proximal interphalangeal joints Feet - Metatarsus varus - Talipes equinovarus (32%) MUSCLE, SOFT TISSUES - Hypoplastic calf muscles NEUROLOGIC Central Nervous System - Motor developmental delay MOLECULAR BASIS - Caused by mutation in the fibrillin 2 gene (FBN2, 121050.0001 ) ▲ Close
Congenital contractual arachnodactyly Other names Beals syndrome; Beals–Hecht syndrome; Arachnodactyly, contractural Beals type; multiple with arachnodactyly; Ear anomalies-contractures-dysplasia of bone with kyphoscoliosis; Distal arthrogryposis type 9 Symptoms Tall, slender body; arm span exceeds height; long, slender fingers and toes; kyphoscoliosis; crumpled ear; joint stiffness Usual onset Conception Causes Mutation of FBN2 gene Treatment Physical therapy for joint contractures; bracing and/or surgical correction for kyphoscoliosis Prognosis Life expectancy depends on severity of symptoms but typically it is not shortened Congenital contractural arachnodactyly ( CCA ), also known as Beals-Hecht syndrome , is a rare autosomal dominant congenital connective tissue disorder. [1] As with Marfan syndrome , people with CCA typically have an arm span that is greater than their height and very long fingers and toes . [2] However, Beals and Hecht discovered in 1972 that, unlike Marfan's, CCA is caused by mutations to the fibrillin-2 ( FBN2 ) gene rather than the fibrillin-1 ( FBN1 ) gene. [1] [3] Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 4 Management 5 Prognosis 6 See also 7 References 8 External links Signs and symptoms [ edit ] CCA is characterized by contractures of varying degrees, mainly involving the large joints, which are present in all affected children at birth. [1] The contractures may be mild and tend to improve over time, but permanently bent fingers and toes ( camptodactyly ) are almost always present. [1] [4] In addition to long fingers and toes and a tall, slender body, people with CCA often have ears that appear to be crumpled , joint stiffness and underdeveloped muscles (muscular hypoplasia ), and they may have curved spines (congenital kyphoscoliosis ). [1] [2] If kyphoscoliosis is present, it often becomes progressively worse and may require surgery. [2] [5] In some cases, the blood vessel that distributes blood from the heart to the rest of the body ( aorta ) may be abnormally enlarged ( aortic root dilatation ). [4] Causes [ edit ] Congenital contractural arachnodactyly may be the result of new mutations in the FBN2 gene or it may be inherited from a parent in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. [2] Congenital contractural arachnodactyly is inherited in an autosomal dominant pattern.
CCA is caused by mutations in a gene called FBN2 gene and is inherited in an autosomal dominant pattern . CCA shares similiar signs and symptoms to Marfan syndrome ; however, Marfan syndrome is not caused by mutations in the FBN2 gene.
Congenital contractural arachnodactyly (CCA, Beals syndrome) is a connective tissue disorder characterized by multiple flexion contractures, arachnodactyly, severe kyphoscoliosis, abnormal pinnae and muscular hypoplasia. Epidemiology The incidence of CCA is unknown and its prevalence is difficult to estimate due to the overlap in phenotype with MFS. Etiology Beals syndrome is caused by a mutation in the FBN2 gene on chromosome 5q23. The number of patients reported has increased following the identification of the FBN2 mutation. Diagnostic methods Ultrasound imaging may be used to demonstrate joint contractures and hypokinesia in suspected cases.
Description Mycosis fungoides is a malignant T-cell lymphoma of the skin, first reported (and named) by Alibert (1835). ... Greene et al. (1982) found that of 526 consecutive patients with cutaneous T-cell lymphomas, 21 had first-degree relatives with lymphoproliferative or hematopoietic malignancies--29 such malignancies in 21 kindreds. ... Da Silva Almeida et al. (2015) performed whole-exome sequencing of tumor-normal sample pairs from 25 patients with Sezary syndrome and 17 patients with other cutaneous T cell lymphomas (CTCLs).
Mycosis fungoides is the most common form of a type of blood cancer called cutaneous T-cell lymphoma. Cutaneous T-cell lymphomas occur when certain white blood cells, called T cells , become cancerous; these cancers characteristically affect the skin, causing different types of skin lesions. Although the skin is involved, the skin cells themselves are not cancerous. Mycosis fungoides usually occurs in adults over age 50, although affected children have been identified. Mycosis fungoides may progress slowly through several stages, although not all people with the condition progress through all stages.
Sézary syndrome is an aggressive form of a type of blood cancer called cutaneous T-cell lymphoma. Cutaneous T-cell lymphomas occur when certain white blood cells, called T cells, become cancerous; these cancers characteristically affect the skin, causing different types of skin lesions. In Sézary syndrome, the cancerous T cells, called Sézary cells, are present in the blood, skin, and lymph nodes. A characteristic of Sézary cells is an abnormally shaped nucleus, described as cerebriform. People with Sézary syndrome develop a red, severely itchy rash (erythroderma) that covers large portions of their body.
Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This section's tone or style may not reflect the encyclopedic tone used on Wikipedia . ... History [ edit ] Mycosis fungoides was first described in 1806 by French dermatologist Jean-Louis-Marc Alibert . [21] [22] The name mycosis fungoides is very misleading—it loosely means "mushroom-like fungal disease". ... ^ a b c Harvey, Nathan T.; Spagnolo, Dominic V.; Wood, Benjamin A. (2015-12-01). " ' Could it be mycosis fungoides?'
Classical MF predominantly affects adults and the elderly (median age at diagnosis: 55-60 years). Clinical description The disease first manifests by skin lesions consisting of flat patches, preferentially located asymmetrically on the buttocks and other sun-protected areas (lower trunk and thighs, and the breasts in women).
Mycosis fungoides is a disease in which T-cell lymphocytes (a type of white blood cell) become malignant (cancerous) and affect the skin. This condition is one of the most common types of T-cell lymphoma . Mycosis fungoides is characterized by a scaly, red rash that develops on the skin, particularly on areas that are not usually exposed to the sun. The rash may last for months or years without causing any symptoms. Over time, a thin, reddened, eczema-like rash may develop, followed by thickened, red patches of skin. Finally, tumors form which may develop into ulcers and become infected. Mycosis fungoides is difficult to cure. Treatment is usually palliative, with the intention of relieving symptoms and improving the quality of life.
Clinical Features Makrythanasis et al. (2016) reported 5 patients from 3 unrelated families with intellectual disability, hypotonia, and early-onset seizures. In the first family, 2 sibs presented at birth with severe hypotonia and hyporeflexia and later developed resistant seizures at age 4 months. ... The mutations, which were found by whole-exome sequencing and confirmed by Sanger sequencing, segregated with the disorder in the families. ... INHERITANCE - Autosomal recessive GROWTH Other - Intrauterine growth retardation (in some patients) - Poor growth (in some patients) MUSCLE, SOFT TISSUES - Hypotonia, severe NEUROLOGIC Central Nervous System - Delayed psychomotor development - Poor or absent speech - Seizures - Ataxia (in 1 family) - Thin corpus callosum (in family C) - Cerebellar hypoplasia (in family C) - Cerebral atrophy (in family C) Peripheral Nervous System - Hyporeflexia MISCELLANEOUS - Onset at birth - Onset of seizures in first year of life - Five patients from 3 unrelated families have been reported (last curated April 2016) MOLECULAR BASIS - Caused by mutation in the phosphatidylinositol glycan anchor biosynthesis class G protein gene (PIGG, 616918.0001 ) ▲ Close
A rare congenital disorder of glycosylation characterized by early onset of hypotonia, severe global developmental delay, intellectual disability, and seizures. Ataxia, mild facial dysmorphism, and autistic behavior have also been reported. Brain MRI findings are variable and include cerebral atrophy, cerebellar hypoplasia/atrophy, and thin corpus callosum.
This means acute inflammation can be broadly divided into a vascular phase that occurs first, followed by a cellular phase involving immune cells (more specifically myeloid granulocytes in the acute setting). ... Plasma cascade systems [ edit ] The complement system , when activated, creates a cascade of chemical reactions that promotes opsonization , chemotaxis , and agglutination , and produces the MAC . ... Emerging evidence now suggests that an active, coordinated program of resolution initiates in the first few hours after an inflammatory response begins. After entering tissues, granulocytes promote the switch of arachidonic acid –derived prostaglandins and leukotrienes to lipoxins, which initiate the termination sequence. ... Chronic inflammation is characterised by the dominating presence of macrophages in the injured tissue.
The relatively recent novel vector has facilitated a far more rapid spread than the simple expansion of habitats north through global warming. [12] In August 2006, cases of bluetongue were found in the Netherlands, then Belgium, Germany, and Luxembourg. [13] [14] In 2007, the first case of bluetongue in the Czech Republic was detected in one bull near Cheb at the Czech-German border. [15] In September 2007, the UK reported its first ever suspected case of the disease, in a Highland cow on a rare-breeds farm near Ipswich, Suffolk . [16] Since then, the virus has spread from cattle to sheep in Britain. [17] By October 2007, bluetongue had become a serious threat in Scandinavia and Switzerland [18] and the first outbreak in Denmark was reported. [19] In autumn 2008, several cases were reported in the southern Swedish provinces of Småland, Halland, and Skåne, [20] as well as in areas of the Netherlands bordering Germany, prompting veterinary authorities in Germany to intensify controls. [21] Norway had its first finding in February 2009, when cows at two farms in Vest-Agder in the south of Norway showed an immune response to bluetongue. [22] Norway have since been declared free of the disease in 2011. ... However infection at an intermediate stage, before the fetal immune system is fully developed, may result in a chronic infection that lingers until the first months after birth of the lamb. ... He also created the first bluetongue vaccine, which was developed from an attenuated BTV strain. [29] For many decades bluetongue was thought to be confined to Africa. The first confirmed outbreak outside of Africa occurred in Cyprus in 1943. [28] Related diseases [ edit ] African horse sickness is related to bluetongue and is spread by the same midges ( Culicoides species). ... WAHID Interface – OIE World Animal Health Information Database Disease card UK government page from Defra Bluetongue page on warmwell.com Canadian Food Inspection Agency (CFIA) Animal Disease Information Bluetongue disease fact sheet Biosecurity training video Farm-level biosecurity practices Takamatsu, H; Mellor, PS; Mertens, PP; Kirkham, PA; Burroughs, JN; Parkhouse, RM (January 2003).
Clinical Features The disorder was first described by Leri and Weill (1929). ... Dawe et al. (1982) reviewed 13 patients with dyschondrosteosis from 8 families and concluded that inheritance is likely to be autosomal dominant, but with only 50% penetrance. ... Roubicek et al. (2003) suggested that since the SHOX gene is located in the pseudoautosomal region of the sex chromosomes, the correct term for the type of hereditary transmission of the associated phenotype should be a 'pseudoautosomal dominant,' 'sex chromosomal dominant,' or 'gonosomal dominant' form of mesomelic dysplasia. ... In a Thai family with an autosomal dominant skeletal dysplasia with similarities to dyschondrosteosis, referred to as the Kantaputra type of mesomelic dysplasia (MDK; 156232), Fujimoto et al. (1998) demonstrated linkage to markers in the 2q24-q32 region. ... Hands and feet were normal. There was fusion between the first and second cervical vertebrae and a cleft in the vertebrae in the lumbosacral region.
Genetic counseling LWD is inherited in a pseudoautosomal dominant manner with each child of an affected individual having a 50% risk of inheriting the mutation.
Onoufriadis et al. (2012) analyzed the RNF114 gene in 485 psoriasis patients and 842 controls and identified 4 disease-associated variants in the RNF114 promoter, each of which was present in a single patient.
Most insulinomas are small, less than 2 cm. [ citation needed ] History [ edit ] Hypoglycemia was first recognized in the 19th century. ... A pioneering description of hyperinsulinism as a cause of hypoglycemia was published by Seale Harris in 1924. The first report of a surgical cure of hypoglycemia by removing an islet cell tumour was in 1929. An insulinoma removed from a woman in Munich provided insulin mRNA that was used in the first human gene cloning experiment. In 1979, Axel Ulrich cloned this gene into E. coli . ... PMID 22198808 . S2CID 7329783 . ^ MeSH website , tree at: "Pancreatic Neoplasms [C04.588.322.475]", accessed 16 October 2014 ^ Sotoudehmanesh R, Hedayat A, Shirazian N, Shahraeeni S, Ainechi S, Zeinali F, Kolahdoozan S (June 2007).
Insulinoma is a type of pancreatic neuroendocrine tumor (pancreatic NET), which refers to a group of rare tumors that form in the hormone-making cells of the pancreas. Insulinomas, specifically, produce too much insulin, a hormone that reduces the level of sugar in the blood by helping it move into cells. As a result, people with insulinomas generally have very low blood sugar levels which can be associated with anxiety, confusion, hunger, a fast heart rate, and sweating. In severe cases, it can lead to seizures, coma or even death. Ninty percent of insulinomas are benign (noncancerous). In most cases, the underlying cause of insulinoma is unknown. However, people with specific genetic syndromes such as multiple endocrine neoplasia type I , Von Hippel-Lindau syndrome , Neurofibromatosis type 1 , and tuberous sclerosis are at risk of insulinomas and other endocrine tumors .
Genetic counseling Although the majority of insulinomas occur sporadically, about 5% to 10% may be associated with MEN1, which is inherited in an autosomal dominant manner. Genetic counseling should be offered to insulinoma patients with MEN1.
Symptoms In general, the symptoms of postherpetic neuralgia are limited to the area of skin where the shingles outbreak first happened. That's commonly in a band around the trunk of the body, most often on one side. ... When to see a doctor See a health care provider at the first sign of shingles. Often the pain starts before you notice a rash. ... But you should use only a small amount when you first try it to make sure you don't have bad side effects. ... Are there brochures or other printed material I can have? What websites do you recommend? Feel free to ask other questions too.
"PHN" redirects here. For other uses, see PHN (disambiguation) . Postherpetic neuralgia Specialty Neurology Symptoms burning or stabbing pain, pain doesn't end after the shingles subsides. Duration lifelong Postherpetic neuralgia ( PHN ) is neuropathic pain that occurs due to damage to a peripheral nerve caused by the reactivation of the varicella zoster virus ( herpes zoster , also known as shingles ). Typically, the nerve pain (neuralgia) is confined to an area of skin innervated by a single sensory nerve , which is known as a dermatome . PHN is defined as dermatomal nerve pain that persists for more than 90 days after an outbreak of herpes zoster affecting the same dermatome. [1] Several types of pain may occur with PHN including continuous burning pain, episodes of severe shooting or electric-like pain, and a heightened sensitivity to gentle touch which would not otherwise cause pain (mechanical allodynia ) or to painful stimuli ( hyperalgesia ). [1] Abnormal sensations and itching may also occur. [1] The nerve pain of PHN is thought to result from damage in a peripheral nerve that was affected by the reactivation of the varicella zoster virus or troubles after chemotherapy. PHN typically begins when the herpes zoster vesicles have crusted over and begun to heal, but can begin in the absence of herpes zoster—a condition called zoster sine herpete . [ citation needed ] There is no treatment that modifies the disease course of PHN; therefore, controlling the affected person's symptoms is the main goal of treatment.