Joubert syndrome with ocular defect is, along with pure JS, the most frequent subtype of Joubert syndrome and related disorders (JSRD, see these terms) characterized by the neurological features of JS associated with retinal dystrophy.
A number sign (#) is used with this entry because of evidence that Joubert syndrome-3 (JBTS3) is caused by homozygous mutation in the AHI1 gene (608894) on chromosome 6q23. For a phenotypic description and a discussion of genetic heterogeneity of Joubert syndrome, see JBTS1 (213300). Clinical Features Lagier-Tourenne et al. (2004) described 2 consanguineous families with Joubert syndrome, one Turkish and the other of Swiss origin; the latter was originally described by Boltshauser and Isler (1977). ... In affected members of 3 consanguineous families with Joubert syndrome, some with cortical polymicrogyria, Dixon-Salazar et al. (2004) identified 1 missense and 2 frameshift mutations in the AHI1 gene. ... In addition to the molar tooth sign, retinal dystrophy was present in 12 families; however, no individuals exhibited variable signs of Joubert syndrome such as polydactyly, encephalocele, colobomas, or liver fibrosis. ... A phenotype-specific group of Joubert syndrome plus retinopathy had an AHI1 mutation frequency was 21.7% (5 of 23 probands).
Description Oculopalatocerebral syndrome is a rare disorder characterized by low birth weight, microcephaly, persistent hyperplastic primary vitreous, microphthalmia, large ears, small hands and feet, cleft palate, joint hypermobility, developmental delay, and cerebral atrophy (summary by Pellegrino et al., 2001). ... Alanay et al. (2004) described a third patient with oculopalatocerebral syndrome, supporting autosomal recessive inheritance because of parental consanguinity. ... In the family reported by Alanay et al. (2004), a second case of oculopalatocerebral syndrome had occurred. This was the son of a paternal uncle married to a second cousin. ... Alanay et al. (2004) suggested that the disorder should be called oculopalatocerebral syndrome rather than oculopalatocerebral dwarfism, as short stature may not be a feature. Inheritance Parental consanguinity in the patients with oculopalatocerebral syndrome reported by Frydman et al. (1985) and Alanay et al. (2004) suggests autosomal recessive inheritance.
Oculopalatocerebral syndrome is characterised by the association of four anomalies: intellectual deficit, microcephaly, palate anomalies and ocular abnormalities. ... Differential diagnosis Differential diagnosis should include cerebro-oculo-nasal syndrome (see this term) and other syndromes associated with a persistent hyperplastic primary vitreous.
Clinical Features Weyers ulnar ray/oligodactyly syndrome is characterized by variable ulnar, radial, or fibular ray reduction, single central incisor, and renal, splenic, or cardiac anomalies. ... Turnpenny et al. (1992) reviewed syndromes that included ulnar ray reduction. The disorders described originally by de la Chapelle et al. (1972) and reviewed by Whitley et al. (1986) (256050) and Saito et al. (1989) (228940) are similar to Weyers ulnar ray/oligodactyly syndrome. Inheritance If this is a single disorder, it is most likely inherited in an autosomal dominant pattern with incomplete penetrance and variable expressivity. ... Nomenclature Confusion has been generated by the use of the Weyers eponym with several disorders, including Weyers acrofacial dysostosis (Curry-Hall syndrome; 193530). The use of the term Weyers syndrome (without further qualification) should be discontinued for this reason.
Sternal cleft (SC) is a rare idiopathic congenital thoracic malformation characterized by a sternal fusion defect, that can be complete or partial (either superior or inferior), that is usually asymptomatic in the neonatal period (apart from a paradoxical midline thoracic bulging) but that can lead to dyspnea, cough, frequent respiratory infections and increased risk of trauma-related injury to the heart, lungs and major vessels if left untreated.
Clinical description Foix-Chavany-Marie syndrome (FCMS) can occur at any age and patients present with acute-onset bilateral paresis of the facial, lingual, pharyngeal and masticatory muscles (innervated by the V, VII, IX, X and XII cranial nerves). ... In children, it presents congenitally (bilateral opercular polymicrogyria) or as an acquired disorder due to encephalitis, epilepsy and neurodegenerative disorders. The syndrome is generally sporadic but some familial cases have been described. ... Differential diagnosis Differential diagnoses include syndromes that present with bulbar palsy (amyotrophic lateral sclerosis, myasthenia gravis and Brown-Vialetto-van Laere syndrome; see these terms). Clinical overlap of FCMS with congenital bilateral perisylvian polymicrogyria and Worster-Drought syndrome (see these terms) has been noted, suggesting a possible continuum of these conditions.
Foix–Chavany–Marie syndrome Other names Facio-pharyngo-glosso-masticatory diplegia Operculum (brain) Specialty Neurology Foix-Chavany-Marie Syndrome (FCMS), also known as Bilateral Opercular Syndrome , is a neuropathological disorder characterized by paralysis of the facial, tongue, pharynx, and masticatory muscles of the mouth that aid in chewing. [1] The disorder is primarily caused by thrombotic and embolic strokes, which cause a deficiency of oxygen in the brain. ... Subopercular syndrome (lesions in the subcortical corticobulbar projections only). Unilateral anterior syndrome involving the frontal operculum. Posterior syndrome involving the junction between the frontal and the parietal lobe of the operculum. ... Overweg-Plandsoen, PhD (1995). "Operculum Syndrome: Unusual Feature of Herpes Simplex Encephalitis".
A number sign (#) is used with this entry because of evidence that peeling skin syndrome-3 (PSS3) is caused by mutation in the carbohydrate sulfotransferase-8 (CHST8; 610190) gene on chromosome 19q13. One such family has been reported. Description Peeling skin syndrome-3 is characterized by asymptomatic lifelong and continuous shedding of the stratum corneum of the epidermis. ... For a discussion of genetic heterogeneity of peeling skin syndrome, see PSS1 (270300). Clinical Features Cabral et al. (2012) described a 4-generation consanguineous Pakistani family with peeling skin syndrome. ... Inheritance The consanguineous Pakistani family described by Cabral et al. (2012) segregated autosomal recessive peeling skin syndrome. Mapping By SNP genomic mapping and microsatellite linkage analysis, Cabral et al. (2012) mapped PSS3 with a lod score of 10.9 to a 16-Mb region on chromosome 19q13 flanked by markers D19S414 and D19S412.
Peeling skin syndrome (PSS) type A is a non inflammatory form of generalized PSS (see this term), a type of ichthyosis (see this term), characterized by generalized white scaling and superficial painless peeling of the skin. ... Differential diagnosis Differential diagnosis includes other forms of PSS and staphylococcal scalded skin syndrome (see these terms). Antenatal diagnosis The disease is not severe enough to justify prenatal screening.
A number sign (#) is used with this entry because of evidence that peeling skin syndrome-6 (PSS6) is caused by homozygous mutation in the FLG2 gene (616284) on chromosome 1q21. Description Peeling skin syndrome-6 is characterized by generalized ichthyotic dry skin and bullous peeling lesions on the trunk and limbs at sites of minor trauma. ... For a discussion of genetic heterogeneity of peeling skin syndrome, see PSS1 (270300). Clinical Features Alfares et al. (2017) reported a 13-year-old Saudi girl who had skin dryness and peeling from birth, with bullous skin eruptions on the trunk and flexor surfaces of the arms, generally at the site of trauma or contact with hard objects, including shoes. ... Mohamad et al. (2018) studied an affected sister and brother from a consanguineous Arab Moslem family with peeling skin syndrome that primarily involved the limbs as well as the trunk, with sparing of the face and palmoplantar skin. ... Molecular Genetics In 3 affected sibs from a consanguineous Saudi family with peeling skin syndrome, who were negative for mutations in known PSS-associated genes, Alfares et al. (2017) performed exome sequencing and identified homozygosity for a nonsense mutation in the FLG2 gene (S211X; 616284.0001).
Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Specialty Dermatology Causes Deletion in the POMP gene Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome ( KLICK syndrome ) is a rare cutaneous condition characterized by ichthyosis and keratoderma . [1] [2] It is an autosomal recessive disorder associated with a deletion in the transcription gene POMP , which codes proteasome maturation protein. [3] [4] This prevents the correct formation of filaggrin from profilaggrin. [5] Sympotmatic treatment with keratolytics and retinoids is successful, but if treatment is stopped, symptoms recur. [5] See also [ edit ] CEDNIK syndrome List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... PMID 20226437 . ^ Baeta, IG; Pereira, AC; Guedes, AC; Pereira, LB (2011). "Do you know this syndrome?" . Anais Brasileiros de Dermatologia . 86 (3): 605–7. doi : 10.1590/S0365-05962011000300036 .
Vahlquist et al. (1997) described an isolated case of what was thought to be the same disorder, which they referred to as KLICK syndrome (keratosis linearis with ichthyosis congenita and sclerosing keratoderma). ... Inheritance Vahlquist et al. (1997) suggested that KLICK syndrome is an autosomal recessive disorder. ... Mapping Dahlqvist et al. (2010) performed whole-genome SNP analysis on DNA from 3 Spanish sibs and 3 Swedish sporadic cases with KLICK syndrome and identified a 1.5-Mb homozygous candidate region on chromosome 13q. ... Molecular Genetics In 12 patients from 8 European families with KLICK syndrome mapping to chromosome 13q, Dahlqvist et al. (2010) analyzed candidate genes and identified homozygosity for a 1-bp deletion in the POMP gene (613386.0001). ... Immunohistochemical staining of patient skin biopsies revealed an altered distribution of POMP and proteasome subunits during formation of the horny layer, suggesting that KILCK syndrome is caused by proteasome insufficiency at a specific stage of epidermal differentiation.
Keratosis linearis-ichthyosis congenita-sclerosing keratoderma syndrome is an inherited epidermal disorder characterized by palmoplantar keratoderma, linear hyperkeratotic papules on the flexural side of large joints (cord-like distribution around wrists, in antecubital and popliteal folds), hyperkeratotic plaques (on neck, axillae, elbows, wrists, and knees), mild ichthyosiform scaling, and sclerotic constrictions around fingers that present flexural deformities.
MERRF syndrome Other names Fukuhara syndrome "ragged red fibers" in MERRF syndrome Specialty Neurology MERRF syndrome (or myoclonic epilepsy with ragged red fibers ) is a mitochondrial disease . ... It has been observed that patients with MERRF syndrome will primarily display myoclonus as a first symptom. ... Mechanism [ edit ] The mechanism by which MERRFs syndrome occur is not yet well understood. ... "A novel point mutation in the mitochondrial tRNA(Ser(UCN)) gene detected in a family with MERRF/MELAS overlap syndrome". Biochem. Biophys. Res. Commun . 214 (1): 86–93. doi : 10.1006/bbrc.1995.2260 . ... "A novel mitochondrial tRNAPhe mutation causes MERRF syndrome". Neurology . 62 (11): 2119–21. doi : 10.1212/01.wnl.0000127608.48406.f1 .
Features of the MERRF syndrome have also been associated with mutation in the MTND5 gene (516005). ... Her son, who also carried the mutation, had MERRF syndrome; the mother had no signs of MERRF syndrome. ... In a mother and daughter with MERFF/MELAS overlap syndrome, Nakamura et al. (1995) identified a heteroplasmic mutation in the MTTS1 gene (590080.0001). ... The clinical picture was consistent with MELAS syndrome. At age 25 years, he developed myoclonus and ataxia, suggesting progression to MERRF syndrome. ... Mancuso et al. (2004) reported an Italian woman with MERRF syndrome who experienced panic attacks at age 11 years.
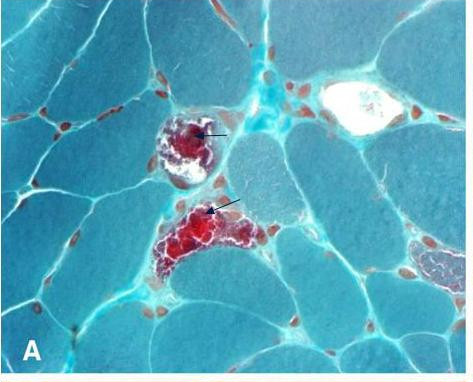
Myoclonic epilepsy with ragged-red fibers (MERRF) is a disorder that affects many parts of the body, particularly the muscles and nervous system. In most cases, the signs and symptoms of this disorder appear during childhood or adolescence. The features of MERRF vary widely among affected individuals, even among members of the same family. MERRF is characterized by muscle twitches (myoclonus), weakness (myopathy), and progressive stiffness (spasticity). When the muscle cells of affected individuals are stained and viewed under a microscope, these cells usually appear abnormal.
Common findings are ptosis, hearing loss, short stature, optic atrophy, cardiomyopathy, cardiac dysrhythmias such as Wolff-Parkinson-White syndrome, and peripheral neuropathy. Pigmentary retinopathy, optic neuropathy, diabetes mellitus, and lipomatosis have been observed. ... Individuals with the diagnosis of Ramsay Hunt syndrome should be investigated for MERRF. ... The differential diagnosis includes other mitochondrial disorders (see Mitochondrial Disorders Overview), syndromes characterized by ataxia (see Hereditary Ataxia Overview) and myoclonus epilepsy (e.g., Unverricht-Lundborg disease, Lafora type progressive myoclonus epilepsy, neuronal ceroid lipofuscinosis, and sialidosis [Kälviäinen 2015]), and the disorders summarized in Table 4. ... Genes of Interest in the Differential Diagnosis of MERRF View in own window Gene(s) DiffDx Disorder MOI Clinical Features of DiffDx Disorder Distinguishing Features CARS2 Combined oxidative phosphorylation deficiency 27 (OMIM 616672) AR Juvenile-onset MERRF-like severe myoclonus epilepsy w/ataxia, spastic tetraparesis, vision loss, hearing loss, & cognitive decline AR inheritance MT-ND5 MT-TC MERRF/MELAS overlap syndrome Mat May initially resemble MERRF 1 Stroke-like episodes MT-TL2 MT-TL2 disorder Mat Features of MERRF & NARP in 1 person 2 Retinitis pigmentosa POLG POLG -related disorders AR Myoclonus, epilepsy, ataxia, peripheral neuropathy Absence of RRF in POLG phenotype AR = autosomal recessive; DiffDx = differential diagnosis; Mat = maternal; MOI = mode of inheritance; NARP = neuropathy-ataxia-retinitis pigmentosa; RRF = ragged red fibers 1. ... Martín-Jiménez et al [2012] Lipomas. Other syndromes that cause multiple lipomas (e.g., multiple symmetric lipomatosis [OMIM 151800]) need to be considered.
Other common findings include hearing loss, short stature, optic atrophy , and cardiomyopathy with Wolff-Parkinson-White (WPW) syndrome . The diagnosis is based on clinical features and a muscle biopsy finding of ragged red fibers (RRF).
MERRF (Myoclonic Epilepsy with Ragged Red Fibers) syndrome is a mitochondrial encephalomyopathy characterized by myoclonic seizures. ... Over 80% of individuals with MERRF syndrome carry the 8344A>G mutation in the lysine transfer RNA ( tRNA Lys ) gene ( MTTK ). ... They may be associated with MERRF/MELAS overlap syndrome, in which affected individuals also suffer from stroke-like episodes. ... Genetic counseling The heteroplasmy makes genetic counseling very arduous in MERRF syndrome. Mitochondrial DNA mutations are transmitted through maternal inheritance. ... Prognosis The prognosis for patients with MERRF syndrome is globally poor because of the progressive nature of the disease.
A specific change in the MTTK gene causes a condition characterized by weakened heart muscle (cardiomyopathy) and hearing loss. Affected individuals may also have myopathy and ataxia. This mutation replaces the DNA building block (nucleotide) guanine with the nucleotide adenine at position 8363 (written as G8363A) within the gene. It is unclear how this alteration in the MTTK gene results in cardiomyopathy, hearing loss, and other symptoms.
Opsoclonus myoclonus syndrome Other names Dancing eye syndrome [1] Specialty Neurology Opsoclonus myoclonus syndrome ( OMS ), also known as opsoclonus-myoclonus-ataxia (OMA), is a rare neurological disorder of unknown cause which appears to be the result of an autoimmune process involving the nervous system . ... Other names for OMS include: [ citation needed ] Opsoclonus-Myoclonus-Ataxia (OMA) Paraneoplastic Opsoclonus-Myoclonus Ataxia (POMA) Kinsbourne syndrome Myoclonic Encephalopathy of Infants Dancing Eyes-Dancing Feet syndrome Dancing Eyes syndrome (see also, Nystagmus ) References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. ... "Opsoclonus-myoclonus syndrome: A clinicopathological confrontation". ... "Delayed, recurrent opsoclonus-myoclonus syndrome responding to plasmapheresis". ... "B- and T-cell markers in opsoclonus-myoclonus syndrome: immunophenotyping of CSF lymphocytes".
Opsoclonus myoclonus syndrome (OMS) is a rare neuroinflammatory disease of paraneoplastic, parainfectious or idiopathic origin, characterized by opsoclonus, myoclonus, ataxia, and behavioral and sleep disorders.
There are very likely other PMLD forms that have not been defined but that are caused by mutations affecting other genes involved in myelination. Other syndromes have also been referred to as PMLD but their inclusion as PMLD has been debated because of their severity and of the evidence of neuronal, besides white matter, involvement on MRI. These syndromes include an autosomal recessive syndrome due to mutations in the HSPD1 gene, encoding the heat shock protein 1, that resembles severe PMD (nystagmus, developmental delay, spasticity, feeding and breathing problems, early-onset lethality) and that is associated with acquired microcephaly, as well as a syndrome due to mutations in the AIMP1 gene, encoding the aminoacyl tRNA synthetase complex-interacting multifunctional protein 1, and characterized by nystagmus, axial hypotonia, spastic paraparesis, severe developmental delay, kyphoscoliosis, microcephaly, intellectual deficit, and absence of speech. An X-linked syndrome, allelic to Allan-Herndon-Dudley syndrome (see this term), has also been referred to as a PMLD. This syndrome is characterized by neonatal hypotonia, nystagmus, progressive spastic paraplegia, ataxia and developmental delay, and is due to mutations in the SLC16A2 gene encoding the monocarboxylate transporter 8 involved in thyroid hormone transport.
A number sign (#) is used with this entry because of evidence that autosomal recessive hypomyelinating leukodystrophy-3 (HLD3) is caused by homozygous mutation in the AIMP1 gene (603605) on chromosome 4q24. Description Autosomal recessive hypomyelinating leukodystrophy-3 (HLD3) is a severe neurologic disorder characterized by early infantile onset of global developmental delay, lack of development, lack of speech acquisition, and peripheral spasticity associated with decreased myelination in the central nervous system (summary by Feinstein et al., 2010). The disorder is phenotypically similar to X-linked Pelizaeus-Merzbacher disease (PMD; 312080), which is caused by mutation in the PLP1 gene (300401). For a general phenotypic description and a discussion of genetic heterogeneity of HLD, see 312080. Clinical Features Early Descriptions Nisenbaum et al. (1965) described a family in which 6 of 7 sibs (3 males and 3 females) died in the first months of life from an infantile disease resembling Pelizaeus-Merzbacher disease.
Postorgasmic illness syndrome is a rare urogenital disease characterized by the appearance of flu-like symptoms (fever, extreme fatigue, myalgia, itchy burning eyes, nasal congestion/rhinorrhea), as well as mood changes, irritability and concentration, memory and attention difficulties, within a few minutes to a few hours after ejaculation.
Postorgasmic illness syndrome (POIS) is a rare condition in which a person develops flu-like and allergy symptoms after orgasm, whether with a partner, through masturbation, or spontaneously during sleep.
A number sign (#) is used with this entry because trichorhinophalangeal syndrome type I (TRPS1) is caused by heterozygous mutation in the TRPS1 gene (604386) on chromosome 8q23. ... Description Trichorhinophalangeal syndrome type I is a malformation syndrome characterized by distinctive craniofacial and skeletal abnormalities and is inherited as an autosomal dominant (Momeni et al., 2000). ... The daughter, however, showed no features of the trisomy 8 syndrome and was a little more affected than the mother. ... Ludecke et al. (1995) and Hou et al. (1995) presented evidence that the Langer-Giedion syndrome is a contiguous gene syndrome due to loss of functional copies of both the TRPS1 and the EXT1 gene (608177) and that the EXT1 gene is distal to the TRPS1 gene. In contrast to TRPS I patients, most TRPS II (Langer-Giedion syndrome) patients have cytogenetically visible deletions and are often mentally retarded.
Trichorhinophalangeal syndrome type I (TRPS I) is a condition that causes bone and joint malformations; distinctive facial features; and abnormalities of the skin, hair, teeth, sweat glands, and nails. ... This condition, called trichorhinophalangeal syndrome type II (TRPS II), has many of the same signs and symptoms of TRPS I, as well as multiple benign (noncancerous) bone tumors called osteochondromas and intellectual disability. ... Learn more about the gene associated with Trichorhinophalangeal syndrome type I TRPS1 Inheritance Pattern TRPS I is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
A rare genetic disease characterized by sparse scalp hair, lateral thinning of eyebrows, mild facial dysmorphism (bulbous tip of the nose, long flat philtrum, thin upper lip vermilion, and protruding ears), and skeletal anomalies including cone-shaped phalangeal epiphyses, hip dysplasia, and short stature. Type 3 can be differentiated by the presence of severe brachydactyly due to short metacarpals. Cartilaginous exostoses are not present in both types.
A group of rare genetic hypopigmentation disorders characterized by a generalized reduction in pigmentation of hair, skin and eyes and variable ocular findings including nystagmus, reduced visual acuity and photophobia. Variants include OCA1A (the most severe form), OCA1B, OCA1-minimal pigment (OCA1-MP), OCA1-temperature sensitive (OCA1-TS), OCA2, OCA3, OCA4, OCA5, OCA6, OCA7 and OCA8.
Oculocutaneous albinism is a group of conditions that affect the coloring of the hair and eyes. Individuals affected by oculocutaneous albinism have very light skin and light-colored irises ; they may also have vision problems such as decreased sharpness of vision, rapid eye movements (nystagmus), crossed eyes ( strabismus ), or increased sensitivity to light ( photophobia ). All types of oculocutaneous albinism are caused by gene mutations that are inherited in an autosomal recessive manner. Treatment includes covering the skin from sun exposure by using sunscreen and protective clothing and attending to vision problems by wearing glasses.
Surgical correction Fetal warfarin syndrome is a disorder of the embryo which occurs in a child whose mother took the medication warfarin (brand name: Coumadin ) during pregnancy . ... Microphthalmia ; abnormally small eyes, telecanthus ; abnormally far apart eyes and strabismus ; misaligned or crossed eyes are common signs of fetal warfarin syndrome. [6] The appearance of an ectopic lacrimal duct , where the tear duct protrudes laterally onto the eye has also been noted. [6] Bodily features [ edit ] Whole body skeletal abnormalities are common in fetal warfarin syndrome. ... Abnormalities of the chest: either pectus carinatum ; [3] a protruding sternum, or pectus excavatum ; [6] a sunken sternum form an immediately recognizable sign of fetal warfarin syndrome. Congenital heart defects such as a thinned atrial septum , coarctation of the aorta , patent ductus arteriosus ; a connection between the pulmonary artery and aorta occur in 8% of fetal warfarin syndrome patients. ... Prevention [ edit ] Fetal warfarin syndrome is prevented by withholding prescription to pregnant women or those trying to conceive . ... External links [ edit ] Classification D ICD - 10 : Q86.0 ICD - 9-CM : 760.7 MeSH : C536683 DiseasesDB : 33178 Datagenno - Fetal Warfarin Syndrome v t e Congenital malformation due to substance exposure Fetal alcohol spectrum disorder Fetal hydantoin syndrome Fetal warfarin syndrome Prenatal amphetamine exposure Prenatal cannabis exposure Prenatal cocaine exposure Prenatal nicotine exposure Other Substance use disorder
Vitamin K antagonist embryofetopathy is characterized by a group of symptoms that may be observed in a fetus or newborn when the mother has taken oral vitamin K antagonists, such as warfarin during pregnancy. Vitamin K antagonists are anticoagulant drugs that provide efficient thromboprophylaxis and that can cross the placenta. 5-12 % of infants exposed to warfarin between 6-9 weeks gestation present nasal hypoplasia and skeletal abnormalities, including short limbs and digits (brachydactyly), and stippled epiphyses. Warfarin fetopathy with central nervous system abnormalities (hydrocephalus, intellectual disability, spasticity, and hypotonia) or ocular abnormalities (microphthalmia, cataract, optic atrophy), fetal loss, and stillbirth, occurs in infants exposed at later gestations. Additional features that have been reported after in utero warfarin exposure include facial dysmorphism (cleft lip and/or palate, malformed ears), choanal atresia or stenosis, aorta coarctation, situs inversus totalis, bilobed lungs, and ventral midline dysplasia.
Acquired von Willebrand syndrome (AVWS) is a bleeding disorder that can occur due to a variety of conditions, but is not caused by a VWF gene mutation. ... This condition may result from lymphoproliferative disorders, autoimmune disorders (including systemic lupus erythrematosus , scleroderma , and antiphospholipid antibody syndrome ), heart conditions such as aortic valvular stenosis , increased blood platelet count such as essential thrombocythemia , and certain drugs.
Epidemiology Prevalence is unknown, but Acquired von Willebrand syndrome (AVWS) is a rare disease that is underdiagnosed, with just over 300 cases reported in the literature so far.
Ring chromosome 19 syndrome is a rare chromosomal anomaly syndrome with a highly variable phenotype that may range from normal to patients with profound intellectual disability, developmental delay, learning disability (esp. speech) and mild dysmorphism (incl. micro/macrocephaly, prominent forehead, low-set and posteriorly rotated ears, hypertelorism, high nasal bridge, prominent philtrum, retro/micrognathia).
Severe intellectual disability-progressive postnatal microcephaly-midline stereotypic hand movements syndrome is a rare, genetic, syndromic intellectual disability disorder characterized by severe intellectual disability, non-inherited, progressive, post-natal microcephaly, hypotonia, hyperkinesia, absence of speech, strabismus, and midline stereotypic hand movements (e.g. hand washing/rubbing).
Silver-Russell syndrome due to maternal uniparental disomy of chromosome 7 is a genetic malformation syndrome with short stature characterized by severe prenatal and postnatal growth retardation, feeding difficulties, body asymmetry, dysmorphic craniofacial features (triangular-shaped face, relative macrocephaly, frontal bossing, micrognathia, down-turned corners of the mouth) and other anomalies (fifth finger clinodactyly, café au lait macules, male genital anomalies, mild developmental delay and/or speech delay with movement disorders).
X-linked intellectual disability-cardiomegaly-congestive heart failure syndrome is a rare X-linked syndromic intellectual disability disorder characterized by profound intellectual disability, global developmental delay with absent speech, seizures, large joint contractures, abnormal position of thumbs and middle-age onset of cardiomegaly and atrioventricular valve abnormalities, resulting in subsequent congestive heart failure.
A number sign (#) is used with this entry because of evidence that syndromic X-linked mental retardation-32 (MRXS32) is caused by mutation in the CLIC2 gene (300138) on chromosome Xq28. ... Molecular Genetics Using exome capture and deep sequencing of genes on the X chromosome in a family with X-linked syndromic mental retardation, Takano et al. (2012) identified a missense mutation in the CLIC2 gene (H101Q; 300138.0001).