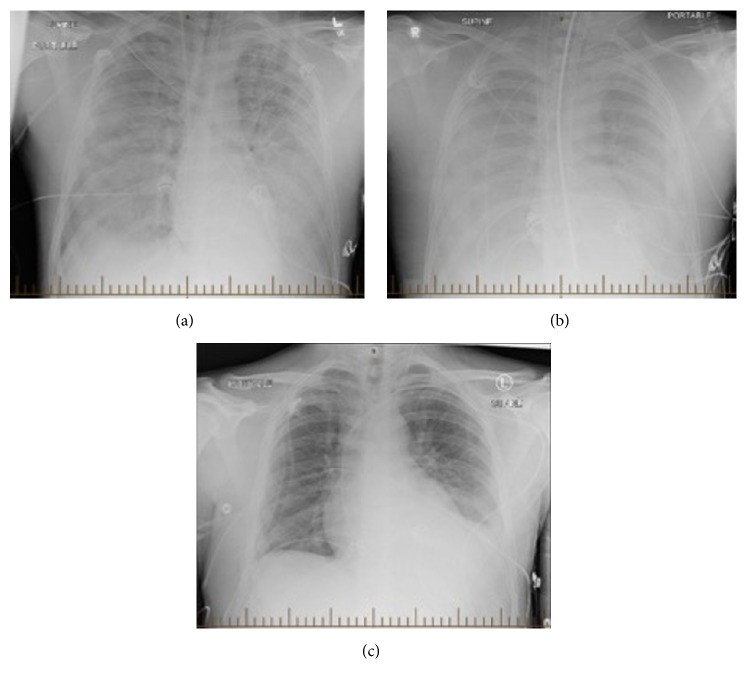
Polyvalvular heart disease syndrome is a recently described syndrome characterized by the combination of polyvalvular heart disease, short stature, facial anomalies and intellectual deficit.
Emery-Nelson syndrome is a rare congenital limb malformation syndrome characterized by facial dysmorphism (high forehead, depressed nasal bridge, long philtrum, flat malar region, high arched palate), short stature and deformities of the hands and feet (small hands/feet, flexion contractures of the first three metacarpophalangeal joints, extension contractures of the thumbs at the interphalangeal joints, clawed toes, mild pes cavus).
Emery and Nelson (1970) reported a mother and daughter with the same disorder. The mother's condition was known by history only. Nonprogressive deformities of the hands were first noted in childhood. The face was flat. Both were about 5 feet tall. The daughter was mentally retarded but the mother was considered unusually intelligent. The daughter was 'floppy' as a neonate. The first three metacarpophalangeal joints had flexion contractures and the thumbs showed contractures in extension at the interphalangeal joints. All the toes were clawed. Limbs - Nonprogressive deformities of hands - Metacarpophalangeal flexion contractures - Interphalangeal extension contractures of thumbs - Clawed toes Growth - Short stature Neuro - Mental retardation - Neonatal hypotonia Facies - Flat face Inheritance - Autosomal dominant ▲ Close
Ring chromosome 2 is a very rare chromosome abnormality in which the ends of chromosome 2 join together to form a ring shape. The severity and symptoms associated with ring chromosome 2 vary from person to person. Slow growth, short stature and a small head size are common features, but people with ring chromosome 2 may otherwise be generally healthy with no major birth defects. Signs and symptoms present in some people may include failure to thrive, developmental delay, low muscle tone (hypotonia), distinctive facial features, skeletal abnormalities, and/or other birth defects involving the heart, genitals, or other parts of the body. Ring chromosome 2 typically is not inherited and occurs sporadically, during the formation of egg or sperm cells or shortly after the egg and sperm join together.
Hypermobile Ehlers-Danlos syndrome is an inherited connective tissue disorder that is caused by defects in a protein called collagen . It is generally considered the least severe form of Ehlers-Danlos syndrome (EDS) although significant complications can occur.
Negative testing for an arterial fragility syndrome also does not confirm a diagnosis of hEDS. ... Marfan syndrome is inherited in an autosomal dominant manner. ... Six genes have been associated with Stickler syndrome. However, a few families with features of Stickler syndrome are not linked to any of these loci; thus, pathogenic variants in other genes may also cause the disorder. Stickler syndrome is diagnosed based on clinical features. ... Fragile X syndrome is inherited in an X-linked manner.
Description The Ehlers-Danlos syndrome shows phenotypic and genetic heterogeneity; see 130000. ... Molecular Genetics Narcisi et al. (1994) reported a family in which multiple members with a connective tissue disorder answering to the description of either EDS III or familial joint instability syndrome (147900) had a mutation in the COL3A1 gene (G637S; 120180.0020).
Hantavirus pulmonary syndrome (HPS) is a severe, respiratory disease caused by infection with a hantavirus. ... Early symptoms universally include fatigue, fever and muscle aches (especially in the thighs, hips, and/or back), and sometimes include headaches, dizziness, chills, and abdominal problems such as nausea, vomiting, diarrhea, and pain. Later symptoms of the syndrome occur 4 to 10 days after initial onset and include coughing and shortness of breath.
A rare viral hemorrhagic fever characterized by virus-induced microvascular leakage rapidly leading to a severe illness with diffuse pulmonary edema and respiratory failure. These symptoms set in after a short first disease stage with fever, myalgia, and headache, followed by severe gastrointestinal symptoms such as abdominal pain, vomiting, and diarrhea. The high lethality of the disease is due to the possible development of hypotension and cardiogenic shock.
Levy H. "Hantavirus cardiopulmonary syndrome: a new twist to an established pathogen", In: Fong IW, editor; Alibek K, editor. ... "Molecular linkage of hantavirus pulmonary syndrome to the white-footed mouse, Peromyscus leucopus: genetic characterization of the M genome of New York virus", J Virol. 1995;69:8137–8141. ... "An unusual hantavirus outbreak in southern Argentina: person-to-person transmission? Hantavirus Pulmonary Syndrome Study Group for Patagonia" . Emerg Infect Dis . 3 (2): 171–4. doi : 10.3201/eid0302.970210 . ... Archived from the original on 2013-10-15. ^ "CDC - Hantavirus Pulmonary Syndrome (HPS) – Hantavirus" . Cdc.gov. 2013-02-06 . Retrieved 2013-07-07 . ^ Akram, Sami (20 November 2020). "Hantavirus Cardiopulmonary Syndrome" . National Center for Biotechnology Information . ^ "Death at the Corners" .
Overview Hantavirus pulmonary syndrome is a rare infectious disease that begins with flu-like symptoms and progresses rapidly to more severe disease. ... The disease is also called hantavirus cardiopulmonary syndrome. Several strains of the hantavirus can cause hantavirus pulmonary syndrome. ... Because treatment options are limited, the best protection against hantavirus pulmonary syndrome is to avoid contact with rodents and safely clean up rodent habitats. ... Causes Rodent carriers Hantavirus pulmonary syndrome is a human disease found only in North and South America. ... Risk factors In the United States, hantavirus pulmonary syndrome is most common in rural areas of the West.
Broken heart syndrome is a temporary condition that affects the heart. ... Signs and symptoms of the condition mimic those of a heart attack and may include sudden chest pain, shortness of breath, and an irregular heartbeat. The cause of broken heart syndrome is not completely understood. Some researchers believe that it may occur when certain hormones released during times of stress temporarily affect the heart's ability to pump blood throughout the body.
X-linked intellectual disability-short stature-overweight syndrome is a multiple congenital anomalies syndrome characterized by borderline to severe intellectual disability, speech delay, short stature, elevated body mass index, a pattern of truncal obesity (reported in older males), and variable neurologic features (e.g. hypotonia, tremors, gait disturbances, behavioral problems, and seizure disorders).
A number sign (#) is used with this entry because of evidence that X-linked mental retardation-12 (MRX12) is caused by mutation in the THOC2 gene (300395) on Xq25. Clinical Features Kumar et al. (2015) reported 4 unrelated families with X-linked mental retardation. Two of the families had previously been reported by Kerr et al. (1992) as 'MRX12' and by Gu et al. (1996) as 'MRX35.' All 20 males in the study of Kumar et al. (2015) had intellectual disability that ranged from borderline to severe. Common additional features included speech delay, short stature, elevated body mass index (BMI), and a truncal obesity pattern in older males.
Nijmegen breakage syndrome-like disorder is a rare, genetic multiple congenital anomalies/dysmorphic syndrome characterized by growth retardation, short stature, developmental delay, intellectual disability, craniofacial dysmorphism (i.e. severe microcephaly, sloping forehead, prominent eyes, broad nasal ridge, hypoplastic nasal septum, epicanthal folds), spontaneous chromosomal instability, cellular hypersensitivity to ionizing radiation and radioresistant DNA synthesis, without severe infections, immunodeficiency or cancer predisposition.
Fibroblasts showed radioresistant DNA synthesis typical of AT or the Nijmegen breakage syndrome (251260). Waltes et al. (2009) reported further phenotyping of this female, who was 23 years of age at that time. ... Molecular Genetics In a patient with a Nijmegen breakage syndrome-like disorder (NBSLD), Waltes et al. (2009) identified compound heterozygosity mutations in the RAD50 gene, a maternally inherited nonsense mutation (604040.0001) and a paternally inherited point mutation that resulted in extension of the protein by 66 amino acids (604040.0002).
The various forms of CFEOM are included in the CCDDs. Other CCDDs include Duane syndrome, Moebius syndrome, and congenital facial palsy. The following conditions can be confused with CFEOM: Brown syndrome ("superior oblique tendon sheath syndrome") is characterized by the inability to elevate the adducted eye actively or passively. ... CHN1 was subsequently found not to be a common cause of sporadic Duane syndrome [Miyake et al 2010]. A contiguous gene deletion syndrome with Duane syndrome is located on 8q13 (DURS1, OMIM 126800). SALL4 -related disorders . The SALL4 -related syndromes include Okihiro syndrome, Duane-radial ray syndrome, acro-renal-ocular syndrome, and IVIC syndrome. ... The three phenotypes are: Kearns-Sayre syndrome (KSS), Pearson syndrome, and progressive external ophthalmoplegia (PEO).
Description Congenital fibrosis of the extraocular muscles (CFEOM) encompasses several different inherited strabismus syndromes characterized by congenital restrictive ophthalmoplegia affecting extraocular muscles innervated by the oculomotor and/or trochlear nerves. ... Clinical Features Khodadoust and von Noorden (1967) described a family in which a bilateral vertical retraction syndrome was present in 2 of 5 sibs. Although ocular motility was normal in the remainder of the family, 1 of the children with the vertical retraction syndrome and 1 without had incomplete situs inversus of the optic nerve head. The clinical features of the vertical retraction syndrome were similar to those of Duane syndrome (126800), although they affected different muscles. ... Tischfield et al. (2010) concluded that these 'TUBB3 syndromes' result from a common defect in axonal guidance during development, which can result in additional neurologic involvement.
A number sign (#) is used with this entry because congenital fibrosis of extraocular muscles type 2 (CFEOM2) is caused by mutations in the ARIX gene (602753) on chromosome 11q13. For a general phenotypic description and a discussion of genetic heterogeneity of various forms of CFEOM, see CFEOM1 (135700) Clinical Features Engle et al. (1997) observed CFEOM in members of 3 consanguineous Saudi Arabian families and demonstrated an autosomal recessive pattern of inheritance. This form of CFEOM differed by showing features suggesting a defect in the superior and inferior divisions of the oculomotor nerve rather than in the superior division, as in CFEOM1. Mapping The locus on chromosome 11q13 is referred to as FEOM2. By genetic linkage analysis of the 3 Saudi Arabian families containing 18 affected individuals, Engle et al. (1997) demonstrated that the recessive form of the disease was not linked to the 'classic' CFEOM (135700) chromosome 12 locus. A genomewide screen using both individual and pooled DNA revealed linkage to markers near the centromere of 11q.
Description Congenital fibrosis of the extraocular muscles (CFEOM) encompasses several different inherited strabismus syndromes characterized by congenital restrictive ophthalmoplegia affecting extraocular muscles innervated by the oculomotor and/or trochlear nerves. ... CFEOM4 (609428), also known as Tukel syndrome, maps to chromosome 21q. CFEOM5 (616219) is caused by mutation in the COL25A1 gene (610004) on chromosome 4q25. ... CFEOM had previously been divided into several clinical entities: general fibrosis syndrome, vertical retraction syndrome, and congenital fibrosis of the inferior rectus. ... In 1 family, affected individuals also had mental disability or mental retardation, which Rudolph et al. (2009) postulated may indicate that the syndrome can be associated with more general neurologic dysfunction in addition to impairment in ocular motility.
Clinical Features Tukel et al. (2005) described 6 affected individuals from a large consanguineous Turkish family who had nonprogressive restrictive ophthalmoplegia with blepharoptosis of the right eye and postaxial oligodactyly/oligosyndactyly of the hands, with the right more severely affected than the left. Mapping Tukel et al. (2005) performed a genomewide scan in the Turkish family with eye and hand anomalies and established linkage to a locus on chromosome 21qter, with a multipoint lod score of 4.53 at D21S1259. Fine mapping defined an approximately 1.5-Mb critical region between D21S1897 and the telomere of the long arm. Inheritance Four affected individuals in the Turkish family reported by Tukel et al. (2005) were offspring of first-cousin marriages, suggesting autosomal recessive inheritance. INHERITANCE - Autosomal recessive HEAD & NECK Head - Compensatory head tilt/chin elevation Eyes - Unilateral (right eye) involvement - Congenital fibrosis of extraocular muscles (CFEOM) - Blepharoptosis - Nonprogressive restrictive ophthalmoplegia (superior rectus, inferior oblique, levator palpebralis dysfunction) SKELETAL Hands - Bilateral postaxial oligodactyly/oligosyndactyly (right side affected greater than left) - Absent carpal bones - Fused carpal bones ▲ Close
The rearrangement was balanced in 3 affected individuals and unbalanced in 1 affected girl who presented with syndromic CFEOM with mental retardation and facial dysmorphism reminiscent of Albright hereditary osteodystrophy-like syndrome (600430), which has been associated with 2qter deletions but does not include ptosis.
A rare syndromic disorder with strabismus characterized by congenital non-progressive ophthalmoplegia affecting the oculomotor and/or trochlear nucleus/nerve and their innervated muscles.
Clinical Features Congenital ocular fibrosis syndrome is a hereditary ocular motility disorder in which restrictive ophthalmoplegia and blepharoptosis are associated with replacement of orbital striated muscle by fibrous tissue (see CFEOM1; 135700). Brodsky et al. (1989) reported a child with congenital ocular fibrosis and oculocutaneous hypopigmentation who also manifested 2 neural misdirection syndromes: synergistic divergence and Marcus Gunn jaw winking. The authors hypothesized that a primary developmental defect involving the establishment of normal neuronal connections might be responsible for this congenital fibrosis syndrome. Brodsky (1998) reported 3 patients, including the patient reported by Brodsky et al. (1989), who had congenital fibrosis syndrome and a variant of synergistic divergence characterized by simultaneous abduction with intorsion and depression of the synkinetically abducting eye.
Congenital fibrosis of extraocular muscles (CFEOM) refers to a group of rare conditions that affect the normal development and function of the muscles that control eye movement and position. In general, people affected by these conditions are unable to move their eyes in certain directions and often have strabismus and/or droopy eyelids (ptosis); however, the severity of the condition and the associated signs and symptoms vary significantly by subtype. CFEOM can be caused by changes (mutations) in several genes, including KIF21A , TUBB3 , PHOX2A , and TUBB2B . In some cases, the underlying genetic cause is unknown. CFEOM can be inherited in an autosomal dominant or autosomal recessive manner, depending on the subtype. Treatment is based on the signs and symptoms present in each person.
The oldest age of diagnosis of nephrotic syndrome among the 46 reported individuals is 18 years [Lovric et al 2017]. ... Genes of Interest in the Differential Diagnosis of Sphingosine Phosphate Lyase Insufficiency Syndrome View in own window Gene(s) DiffDx Disorder(s) MOI Features of the DiffDx Disorder Overlapping w/SPLIS Distinguishing from SPLIS ALDH3A2 Sjögren-Larsson syndrome 1 AR Ichthyosis; intellectual disability; abnormal brain MRI Spasticity; lack of kidney involvement GBA Gaucher disease type 2 AR Ichthyosis; nonimmune hydrops; abnormal brain MRI Hepatosplenomegaly; pancytopenia LAMB2 Pierson syndrome (OMIM 609049) AR Steroid-resistant nephrotic syndrome; developmental delay Microcoria LIPA Lysosomal acid lipase deficiency AR Adrenal calcifications Hepatic fibrosis & cirrhosis; intestinal malabsorption LMX1B Nail-patella syndrome AD Steroid-resistant nephrotic syndrome; sensorineural hearing loss Nail dysplasia; hypoplastic or absent patellae; eye anomalies NPHS1 NPHS2 PLCE1 Congenital nephrotic syndrome types 1, 2, & 3 (OMIM 256300, 600995, 610725) AR Steroid-resistant nephrotic syndrome Absence of additional syndromic findings SMARCAL1 Schimke immunoosseous dysplasia AR Steroid-resistant nephrotic syndrome; T-cell lymphopenia Spondyloepiphyseal dysplasia; numerous lentigines AD = autosomal dominant; AR = autosomal recessive; DiffDx = differential diagnosis; MOI = mode of inheritance; SPLIS = sphingosine phosphate lyase insufficiency syndrome 1. ... Congenital ichthyosis is a feature of several genetic syndromes. Nonsyndromic ichthyosis (e.g., autosomal recessive congenital ichthyosis) can be considered if ichthyosis is the presenting finding; syndromic ichthyosis can be assoc w/Netherton syndrome, Sjögren-Larsson syndrome, & trichothiodystrophy Nonimmune hydrops Enlarged/hemorrhagic adrenals Many genetic conditions are assoc w/fetal hydrops (e.g., chromosome anomalies, RASopathies, & lysosomal storage disorders). 1 Primary adrenal insufficiency Adrenal calcifications, hypothyroidism, cryptorchidism, & micropenis may be assoc w/primary adrenal insufficiency. ... Isolated T-cell deficiency may be found in many primary immune deficiencies (e.g., SCID, 22q11.2 deletion syndrome, CHARGE syndrome, FOXN1 haploinsufficiency, CD3 deficiency, & IL7R deficiency). 3 Combined T-, B-, & NK-cell deficiency can be seen in primary immune deficiencies incl adenosine deaminase deficiency & PNP deficiency. ... It is most often seen in association with severe nephrotic syndrome and rapid progression to end-stage renal disease. 5.
A number sign (#) is used with this entry because of evidence that nephrotic syndrome type 14 (NPHS14) is caused by homozygous or compound heterozygous mutation in the SGPL1 gene (603729) on chromosome 10q21. Description NPHS14 is an autosomal recessive syndromic form of steroid-resistant nephrotic syndrome with multisystemic manifestations. ... For a discussion of genetic heterogeneity of nephrotic syndrome and FSGS, see NPHS1 (256300). Clinical Features Prasad et al. (2017) reported 8 patients from 5 unrelated families with a syndromic disorder characterized by early-onset primary adrenal insufficiency associated often with hyperpigmentation and sometimes with adrenal calcifications. ... Most patients developed steroid-resistant nephrotic syndrome associated with FSGS on biopsy and partial effacement of podocytes on electron microscopy. ... Histologically, FSGS was the main finding, but diffuse mesangial sclerosis was found in cases with congenital nephrotic syndrome. At least 2 unrelated patients had a milder renal phenotype, with onset of nephrotic syndrome in the late teens.
A rare disorder with multisystemic involvement and glomerulopathy characterized by progressive steroid-resistant nephrotic syndrome typically associated with focal segmental glomerulosclerosis, as well as primary adrenal insufficiency with adrenal calcifications.
SUCLA2 -related mtDNA depletion syndrome is inherited in an autosomal recessive manner. ... Plasma MMA level may be more sensitive in identifying SUCLA2 -related mtDNA depletion syndrome than urine organic acid analysis. ... Prevalence SUCLA2 -related mtDNA depletion syndrome is rare; the exact prevalence is unknown. ... Encephalomyopathic mtDNA depletion syndromes present in infancy with hypotonia and global developmental delay. ... Within each phenotypic category, mtDNA depletion syndromes are ordered by relative prevalence. 2.
An autosomal dominant form of Segawa syndrome (128230) is caused by mutation in the GCH1 gene (600225). Description Segawa syndrome is an autosomal recessive neurologic disorder characterized by onset in infancy of dopa-responsive dystonia. ... See also infantile parkinsonism-dystonia syndrome (613135), caused by mutation in the SLC6A3 gene (126455). ... Brautigam et al. (1998) and Wevers et al. (1999) reported 4 unrelated Dutch patients with Segawa syndrome. All had normal pregnancies and deliveries. ... In an Italian boy with a severe form of Segawa syndrome, Brautigam et al. (1999) identified a homozygous mutation in the TH gene (191290.0011).
Tyrosine hydroxylase (TH) deficiency is a rare inherited condition that affects the nervous system . There are three different forms of the condition that vary in severity. The mild form is called TH-deficient dopa-responsive dystonia and typically develops between age twelve months and six years. The two severe forms, which are called infantile parkinsonism and progressive infantile encephalopathy, often begin shortly after birth or during early infancy. Although there is some overlap of features among the three forms, each is associated with unique signs and symptoms.
Summary Clinical characteristics. Tyrosine hydroxylase (TH) deficiency is associated with a broad phenotypic spectrum. Based on severity of symptoms/signs as well as responsiveness to levodopa therapy, clinical phenotypes caused by pathogenic variants in TH are divided into (1) TH-deficient dopa-responsive dystonia (the mild form of TH deficiency), (2) TH-deficient infantile parkinsonism with motor delay (the severe form), and (3) TH-deficient progressive infantile encephalopathy (the very severe form). In individuals with TH-deficient dopa-responsive dystonia (DYT5b, DYT-TH), onset is between age 12 months and 12 years; initial symptoms are typically lower-limb dystonia and/or difficulty in walking. Diurnal fluctuation of symptoms (worsening of the symptoms toward the evening and their alleviation in the morning after sleep) may be present. In most individuals with TH-deficient infantile parkinsonism with motor delay, onset is between age three and 12 months.
Tyrosine hydroxylase (TH) deficiency is a disorder that primarily affects movement, with symptoms that may range from mild to severe. The mild form of this disorder is called TH-deficient dopa-responsive dystonia (DRD). Symptoms usually appear during childhood. Affected individuals may exhibit unusual limb positioning and a lack of coordination when walking or running. In some cases, people with TH-deficient DRD have additional movement problems such as shaking when holding a position (postural tremor) or involuntary upward-rolling movements of the eyes. The movement difficulties may slowly increase with age but almost always get better with medical treatment.
Clinical description Disease presents in infancy (most frequently in the first year of life) with a progressive hypokinetic-rigid syndrome with generalized dystonia, involuntary jerky movements, postural tremor, or gait disturbances that may fluctuate during the day and show good or excellent responsiveness to levodopa (L-dopa) in most cases (>80%). ... Differential diagnoses that need to be considered for the more severe, encephalopathy-like phenotype include febrile infection-related epilepsy syndrome, neonatal hypoxic and ischemic brain injury, other tetrahydrobiopterin (BH4)-related enzyme deficiencies and mitochondrial disorders.
Other less common causes of chiasmal syndrome are metabolic, toxic, traumatic or infectious in nature. ... Systemic hormonal aberrations such as Cushing’s syndrome , galactorrhea and acromegaly usually predate the compressive signs. ... Pathophysiology [ edit ] Lee has divided optic chiasmal syndromes into anterior, middle and posterior locations. [6] Anterior chiasmal syndrome affects the junction of the optic nerve and chiasm. Middle chiasmal syndrome relates to the decussating fibers in the body of the optic chiasm while posterior chiasmal syndrome involves the caudal fibers. ... Visual fields tests will delineate chiasmal syndromes because the missing fields will not cross the midline.
Contents 1 Pathophysiology 1.1 Parinaud syndrome 2 Treatment 3 History 4 See also 5 References 6 Further reading 7 External links Pathophysiology [ edit ] The two different types of near response are caused by different underlying disease processes. ... To settle the question of whether the AR pupil is of central or peripheral origin, it will be necessary to perform iris transillumination (or a magnified slit-lamp examination) in a substantial number of patients who have a pupillary light-near dissociation (with and without tonicity of the near reaction), perhaps in many parts of the world. Parinaud syndrome [ edit ] A third cause of light-near dissociation is Parinaud syndrome , also called dorsal midbrain syndrome. This uncommon syndrome involves vertical gaze palsy associated with pupils that “accommodate but do not react." [5] The causes of Parinaud syndrome include brain tumors (pinealomas), multiple sclerosis and brainstem infarction. ... The near response in tonic pupils is slow and prolonged. See also [ edit ] Adie syndrome Anisocoria Cycloplegia Marcus Gunn pupil Miosis Neurosyphilis Parinaud's syndrome Syphilis References [ edit ] ^ "Light-Near Dissociation :: EHSL - Moran Eye Center Neuro-Ophthalmology Collection" . ... PMID 16845316 . ^ "Convergence Retraction Nystagmus (Parinaud's Syndrome) :: EHSL - Moran Eye Center Neuro-Ophthalmology Collection" .
Frequency 1 in 1,000,000 Deaths 30% mortality rate Febrile infection-related epilepsy syndrome (FIRES) is an epilepsy syndrome in which new-onset refractory status epilepticus (NORSE) is preceded by febrile illness 24 hours to 2 weeks prior to the onset of seizures. The term was previously used for a paediatric syndrome but was redefined to include all ages. [1] FIRES was previously to refer to this syndrome in children aged three to fifteen years old. ... "Febrile Infection-Related Epilepsy Syndrome (FIRES): A Literature Review and Case Study" . ... "Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood". ... "Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood".
A rare, potentially fatal , epileptic encephalopathy characterized by explosive-onset of recurrent multifocal and bilateral tonic-clonic seizures following an unspecific febrile illness. The syndrome develops without a clear acute structural, toxic or metabolic cause, in a patient without previous epilepsy. ... Clinical description Febrile infection-related epilepsy syndrome (FIRES) is most common in school-age children. ... Differential diagnosis Differential diagnoses include, but are not limited to, infectious or autoimmune encephalitis (e.g. anti-NMDAR encephalitis and other antineuronal antibody-related encephalitides, acute disseminated encephalomyelitis), primary angiitis of the central nervous system, acute necrotizing encephalopathy, other infection-induced encephalopathies, metabolic diseases (e.g. mitochondrial disorders, citrillunemia, thiamin metabolism disorders) and genetic epilepsies (e.g. Dravet syndrome, PCDH19 epilepsy). Management and treatment Monitoring in intensive care during the acute phase is mandatory.
For example, carriers of Robertsonian translocations involving chromosome 21 have a higher risk of having a child with Down syndrome . This is known as a 'translocation Downs'. ... Infertility : One of the would-be parents carries a balanced translocation , where the parent is asymptomatic but conceived fetuses are not viable. Down syndrome is caused in a minority (5% or less) of cases by a Robertsonian translocation of the chromosome 21 long arm onto the long arm of chromosome 14 . [7] Chromosomal translocations between the sex chromosomes can also result in a number of genetic conditions, such as XX male syndrome : caused by a translocation of the SRY gene from the Y to the X chromosome By chromosome [ edit ] Overview of some chromosomal translocations involved in different cancers, as well as implicated in some other conditions, e.g. schizophrenia, [8] with chromosomes arranged in standard karyogram order. ... Wikimedia Commons has media related to Chromosomal translocations . v t e Cytogenetics : chromosomes Basic concepts Karyotype Ploidy Genetic material / Genome Chromatin Euchromatin Heterochromatin Chromosome Chromatid Nucleosome Nuclear organization Types Autosome / Sex chromosome (or allosome or heterosome) Macrochromosome/Microchromosome Circular chromosome / Linear chromosome Extra chromosome (or accessory chromosome) Supernumerary chromosome A chromosome/B chromosome Lampbrush chromosome Polytene chromosome Dinoflagellate chromosomes Homologous chromosome Isochromosome Satellite chromosome Centromere position Metacentric Submetacentric Telocentric Acrocentric Holocentric Centromere number Acentric Monocentric Dicentric Polycentric Processes and evolution Mitosis Meiosis Structural alterations Chromosomal inversion Chromosomal translocation Numerical alterations Aneuploidy Euploidy Polyploidy Paleopolyploidy Polyploidization Structures Telomere : Telomere-binding protein ( TINF2 ) Protamine Histone H1 H2A H2B H3 H4 Centromere A B C1 C2 E F H I J K M N O P Q T See also Extrachromosomal DNA Plasmid List of organisms by chromosome count List of sequenced genomes International System for Human Cytogenetic Nomenclature v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22 v t e Mutation Mechanisms of mutation Insertion Deletion Substitution Transversion Transition Mutation with respect to structure Point mutation Nonsense mutation Missense mutation Conservative mutation Silent mutation Frameshift mutation Dynamic mutation Large-scale mutation Chromosomal translocations Chromosomal inversions Mutation with respect to overall fitness Deleterious mutation Advantageous mutation Neutral mutation Nearly neutral mutation Synonymous mutation Nonsynonymous mutation
"The diagnosis and treatment of Munchausen's syndrome". General Hospital Psychiatry . ... ISBN 978-0683301281 . ^ Repper, John (February 1995). "Münchausen syndrome by proxy in health care workers". ... Factitious Disorder/Munchhausen Syndrome. The 5-Minute Clinical Consult. 18th Edition. 2010. ... : untangling the web of Munchausen syndrome, Munchausen by proxy, malingering & factitious disorder . ... "Playing patient, playing doctor: Munchausen syndrome, clinical S/M, and ruptures of medical power".