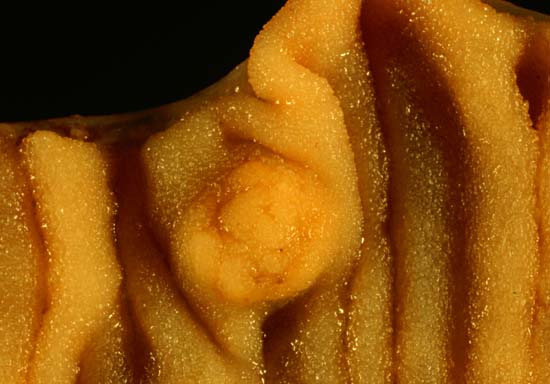
A carcinoid tumor is a type of neuroendocrine tumor that usually develops in the digestive (GI) tract (such as the stomach or intestines) or in the lungs. In some cases, a carcinoid tumor develops in another part of the body, such as the pancreas, testicle (in men), or ovary (in women). It is a slow-growing tumor that typically does not cause symptoms in the early stages, so a person may have the tumor for years before being diagnosed. In later stages, symptoms may vary depending on where the tumor is located. Symptoms of a GI carcinoid tumor may only develop if the tumor has spread to the liver.
Overview Carcinoid tumors are a type of slow-growing cancer that can arise in several places throughout your body. Carcinoid tumors, which are one subset of tumors called neuroendocrine tumors, usually begin in the digestive tract (stomach, appendix, small intestine, colon, rectum) or in the lungs. Carcinoid tumors often don't cause signs and symptoms until late in the disease. Carcinoid tumors can produce and release hormones into your body that cause signs and symptoms such as diarrhea or skin flushing. Treatment for carcinoid tumors usually includes surgery and may include medications.
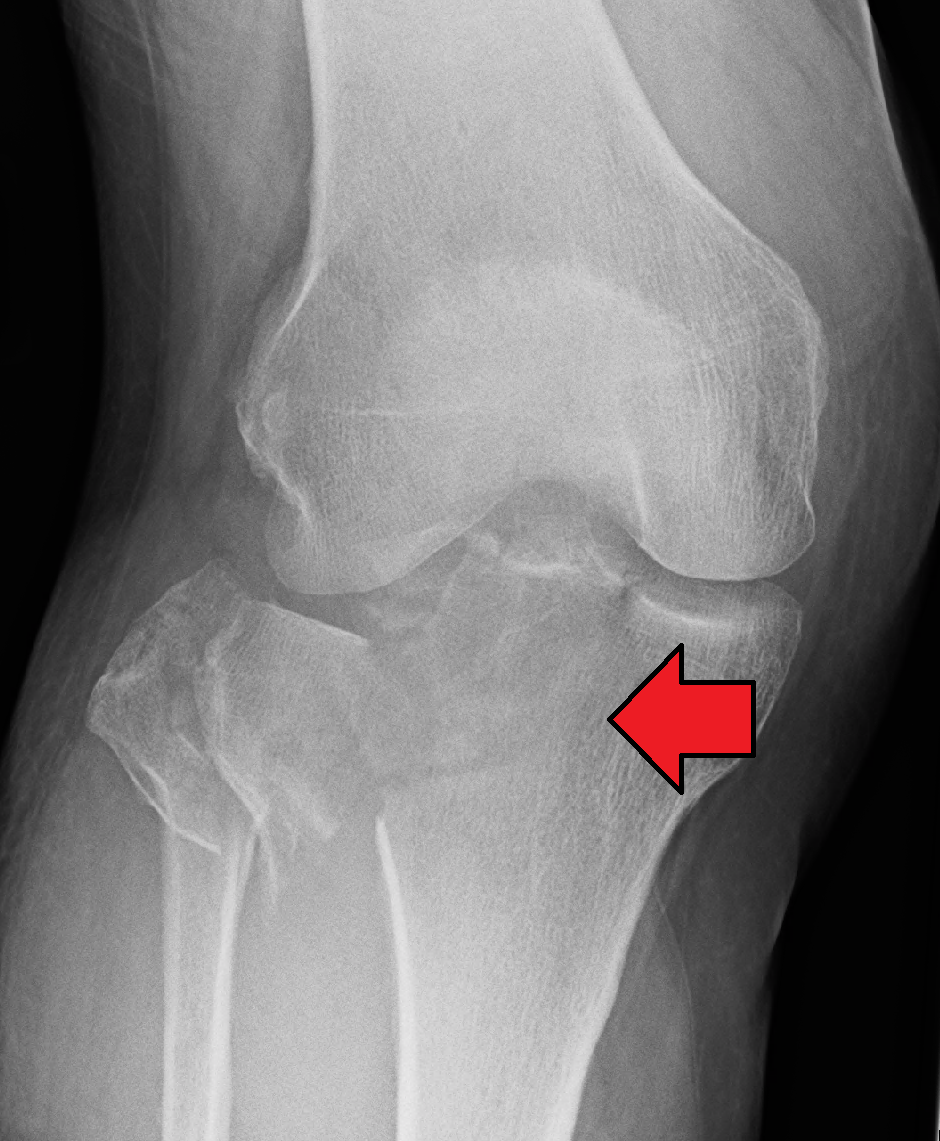
Overview Knee pain is a common complaint that affects people of all ages. Knee pain may be the result of an injury, such as a ruptured ligament or torn cartilage. Medical conditions — including arthritis, gout and infections — also can cause knee pain. Many types of minor knee pain respond well to self-care measures. Physical therapy and knee braces also can help relieve pain. In some cases, however, your knee may require surgical repair. Symptoms The location and severity of knee pain may vary, depending on the cause of the problem.
"Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology". Journal of Psychopharmacology . 28 (5): 403–39. doi : 10.1177/0269881114525674 .
Clinical Features The DSM-IV (American Psychiatric Association, 1994) describes specific phobias, also known as simple phobias, as being characterized by a marked persistent, excessive, and unreasonable fear caused by the presence of a specific object or situation (e.g., flying, heights, animals, injections, blood). Exposure to the phobic stimulus provokes an immediate anxiety response often resembling panic (see panic disorder, 167870). An adult recognizes that the fear is excessive, but a child may not. The phobic situation is either avoided or endured with intense anxiety or distress, and the avoidance or anxiety interferes with the individual's life activities. Vasovagal fainting may occur and is particularly common with blood-injection-injury phobia. Epidemiology studies indicate that women are more likely to have specific phobias than men.
Overview Specific phobias are an extreme fear of objects or situations that pose little or no danger but make you highly anxious. So you try to stay away from these things. Unlike the brief anxiety you may feel when giving a speech or taking a test, specific phobias are long-lasting. Without treatment, specific phobias tend to last a lifetime. Phobias can cause strong physical, mental and emotional responses. They also can affect how you act at work or school, or in social situations. Specific phobias are common anxiety disorders. Overall, they happen more often in females.
Congenital insensitivity to pain is a condition, present from birth, that inhibits the ability to perceive physical pain. Affected individuals are unable to feel pain in any part of their body. Over time, this lack of pain awareness can lead to an accumulation of injuries and health issues that may affect life expectancy. Congenital insensitivity to pain is caused by mutations in the SCN9A gene and, in rare cases, is caused by mutations in the PMRD12 gene . It is inherited in an autosomal recessive pattern. Congenital insensitivity to pain is considered a form of peripheral neuropathy because it affects the peripheral nervous system, which connects the brain and spinal cord to muscles and to cells that detect sensations such as touch, smell, and pain.
Hereditary sensory and autonomic neuropathy type V (HSAN5) is a condition that affects the sensory nerve cells. These cells, which are also called sensory neurons, transmit information about sensations such as pain, temperature, and touch. Signs and symptoms of the condition generally develop at birth or during early infancy and may include a loss of pain and temperature sensation. Because of the inability to feel deep pain, affected people suffer repeated severe injuries such as bone fractures and joint injuries that go unnoticed. HSAN5 is caused by changes (mutations) in the NGF gene and is inherited in an autosomal recessive manner.
A number sign (#) is used with this entry because of evidence that hereditary sensory neuropathy type V (HSAN5) is caused by homozygous mutation in the NGF gene (162030) on chromosome 1p13. For a discussion of genetic heterogeneity of hereditary sensory and autonomic neuropathy, see HSAN1 (162400). Clinical Features Low et al. (1978) reported a 6-year-old child with congenital sensory neuropathy characterized by a selective loss of pain and thermal sensation affecting the extremities. Nerve conduction studies were normal. Small myelinated fibers were selectively reduced in the sural nerve, and unmyelinated fibers were normal. Dyck et al. (1983) reported a girl with congenital insensitivity to pain.
A disorder that is characterized by loss of pain perception and impaired temperature sensitivity, in the absence of any other major neurological anomalies. Epidemiology Prevalence is unknown. Only a small number of cases have been described in the literature, although one large multigenerational consanguineous family from northern Sweden has been reported. Clinical description Other findings include ulcers, self-mutilation and damaged joints. Intelligence is normal. Etiology Mutations in the NGF gene (1p13.1) were detected in affected members of the large Swedish family. However, mutations in the NTRK1 gene (1q21-q22) have been identified in one patient diagnosed with HSAN5 with the additional finding of mild anhidrosis.
National Institute on Drug Abuse . Retrieved 28 October 2018 . ^ Conner, Shayna N.; Bedell, Victoria; Lipsey, Kim; Macones, George A.; Cahill, Alison G.; Tuuli, Methodius G.
"Lymphocytic ("microscopic") colitis: a comparative histopathologic study with particular reference to collagenous colitis". Hum. Pathol . 20 (1): 18–28. doi : 10.1016/0046-8177(89)90198-6 .
Overview Microscopic colitis is an inflammation of the large intestine (colon) that causes persistent watery diarrhea. The disorder gets its name from the fact that it's necessary to examine colon tissue under a microscope to identify it, since the tissue may appear normal with a colonoscopy or flexible sigmoidoscopy. There are different subtypes of microscopic colitis: Collagenous colitis, in which a thick layer of protein (collagen) develops in colon tissue Lymphocytic colitis, in which white blood cells (lymphocytes) increase in colon tissue Incomplete microscopic colitis, in which there are mixed features of collagenous and lymphocytic colitis. Researchers believe collagenous (kuh-LAYJ-uh-nus) colitis and lymphocytic colitis may be different phases of the same condition. Symptoms, testing and treatment are the same for all subtypes. Symptoms Signs and symptoms of microscopic colitis include: Chronic watery diarrhea Abdominal pain, cramps or bloating Weight loss Nausea Fecal incontinence Dehydration The symptoms of microscopic colitis can come and go frequently.
Overview Low sperm count means that the fluid (semen) you ejaculate during an orgasm contains fewer sperm than normal. A low sperm count is also called oligospermia (ol-ih-go-SPUR-me-uh). A complete absence of sperm is called azoospermia. Your sperm count is considered lower than normal if you have fewer than 15 million sperm per milliliter of semen. Having a low sperm count decreases the odds that one of your sperm will fertilize your partner's egg, resulting in pregnancy. Nonetheless, many men who have a low sperm count are still able to father a child.
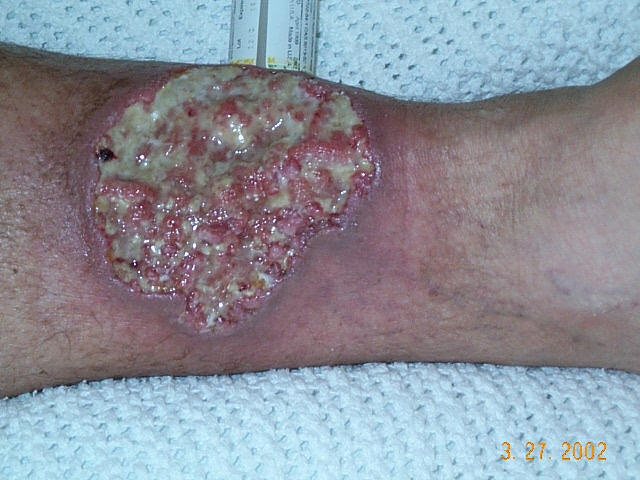
Pyoderma gangrenosum (PG) is a primarily sterile inflammatory neutrophilic dermatosis characterized by recurrent cutaneous ulcerations with a mucopurulent or hemorrhagic exudate. Epidemiology The exact prevalence of PG is unknown. The incidence has been estimated to range between 1 and 3.3 in 330,000. The incidence peak occurs between the ages of 20 to 50 years, with women being more often affected than men. Clinical description Clinically, onset occurs with sterile pustules that rapidly progress and turn into very painful ulcers of variable depth and size, with undermined bluish or violaceous borders and surrounding erythema. The legs are most commonly affected but other parts of the skin and mucous membranes may also be involved.
Overview Pyoderma gangrenosum (pie-o-DUR-muh gang-ruh-NO-sum) is a rare condition that causes large, painful sores (ulcers) to develop on your skin, most often on your legs. The exact causes of pyoderma gangrenosum are unknown, but it appears to be a disorder of the immune system. People who have certain underlying conditions, such as inflammatory bowel disease or arthritis, are at higher risk of pyoderma gangrenosum. Pyoderma gangrenosum ulcers can develop quickly. They usually clear up with treatment, but scarring and recurrences are common. Symptoms Pyoderma gangrenosum usually starts with a small, red bump on your skin, which may resemble a spider bite.
Pyoderma gangrenosum is a rare, destructive inflammatory skin disease of which a painful nodule or pustule breaks down to form a progressively enlarging ulcer. Lesions may occur either in the absence of any apparent underlying disorder or in association with other diseases, such as ulcerative colitis , Crohn's disease , polyarthritis (an inflammation of several joints together), gammopathy , vasculitis, leukemia, and other conditions. Each year in the United States, pyoderma gangrenosum occurs in about 1 person per 100,000 people. Pyoderma gangrenosum belongs to a group of autoinflammatory skin diseases called neutrophilic dermatoses . Neutrophils are a type of white blood cell or leukocyte which form an early line of defense against bacterial infections.
"[Unusual syndrome with acceleration of skeletal maturation (Marshall's syndrome). 1st case in the Italian literature]". Minerva Pediatr. (in Italian). 28 (24): 1499–509. PMID 1012192 . Tzu-Jou Wang (2002).
A number sign (#) is used with this entry because Marshall-Smith syndrome (MRSHSS) is caused by heterozygous mutation in the NFIX gene (164005) on chromosome 19p13. Sotos syndrome-2 (SOTOS2; 614753) is also caused by heterozygous mutation in the NFIX gene. Description The Marshall-Smith syndrome is a malformation syndrome characterized by accelerated skeletal maturation, relative failure to thrive, respiratory difficulties, mental retardation, and unusual facies, including prominent forehead, shallow orbits, blue sclerae, depressed nasal bridge, and micrognathia (Adam et al., 2005). Clinical Features Marshall et al. (1971) described 2 infants with a syndrome characterized by accelerated skeletal maturation, failure to thrive, and dysmorphic facial features. Sperli et al. (1993) reviewed 20 reported cases. Chatel et al. (1998) reported an unusually severe form of Marshall-Smith syndrome characterized by neonatal death.
Marshall-Smith syndrome (MRSHSS) is a genetic disorder in which individuals typically have advanced bone age, difficulties gaining weight (failure to thrive), unique facial features, and intellectual disability. Other signs and symptoms of this condition may include eye abnormalities, breathing difficulties, and neurological issues. Individuals may also have heart defects, an increased amount of body hair (hirsutism), and flat feet (pes planus). MRSHSS is caused by mutations in the NFIX gene. Most individuals with MRSHSS are the first in their family with this condition and are said to have a spontaneous (de novo) mutation. Although there is no specific treatment or cure for MRSHSS, there may be ways to manage the symptoms.
A rare genetic multiple congenital anomalies syndrome characterized by abnormal bone maturation with skeletal anomalies, airway obstructions, failure to thrive, developmental delay, moderate to severe intellectual disability and characteristic facial features with macrocephaly, prominent forehead, shallow orbits, proptosis and blue sclerae. Epidemiology Less than 60 cases have been reported in the literature to date. Clinical description Marshall-Smith syndrome was originally considered as an overgrowth condition based on advanced bone maturation. It is characterized by a dysostosis with skeletal anomalies including progressive kyphoscoliosis, postnatal failure to thrive in weight, short stature, and osteopenia with fractures. Wide bullet-shaped phalanges as well as large hands and feet are observed.
A number sign (#) is used with this entry because pseudovaginal perineoscrotal hypospadias (PPSH) is caused by homozygous or compound heterozygous mutation in the steroid 5-alpha-reductase-2 gene (SRD5A2; 607306) on chromosome 2p23. Description Pseudovaginal perineoscrotal hypospadias is a form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. Clinical Features De Vaal (1955) reported 3 brothers who were thought for a time to be girls. The parents and grandparents on one side were first cousins, and the great-grandparents were also related.
In The Antichrist , Nietzsche applies the word 'idiot' to Jesus in a comparable fashion, almost certainly in an allusion to Dostoevsky's use of the word: [27] "One has to regret that no Dostoevsky lived in the neighbourhood of this most interesting décadent ; I mean someone who could feel the thrilling fascination of such a combination of the sublime, the sick and the childish." [28] [29] References Citations ^ "The Clinical History of 'Moron,' 'Idiot,' and 'Imbecile ' " . merriam-webster.com. ^ a b c Oxford English Dictionary , s.v. ^ a b Liddell-Scott-Jones A Greek–English Lexicon , s.v. ἰδιώτης and ἴδιος . ^ A Latin Dictionary , s.v. ^ du Cange, Glossarium Mediæ et Infimæ Latinitatis , s.v. ^ Trésor de la langue française informatisé , s.v. ^ R.L.
For the common or slang use of imbecile, see Idiot . The term imbecile was once used by psychiatrists to denote a category of people with moderate to severe intellectual disability , as well as a type of criminal. [1] [2] The word arises from the Latin word imbecillus , meaning weak, or weak-minded. [3] It included people with an IQ of 26–50, between " idiot " (IQ of 0–25) and " moron " (IQ of 51–70). [4] In the obsolete medical classification ( ICD-9 , 1977), these people were said to have "moderate mental retardation " or "moderate mental subnormality" with IQ of 35–49. [5] The meaning was further refined into mental and moral imbecility. [6] [7] The concepts of "moral insanity", "moral idiocy"," and "moral imbecility", led to the emerging field of eugenic criminology , which held that crime can be reduced by preventing " feeble-minded " people from reproducing. [8] [9] "Imbecile" as a concrete classification was popularized by psychologist Henry H. Goddard [10] and was used in 1927 by United States Supreme Court Justice Oliver Wendell Holmes Jr. in his ruling in the forced-sterilization case Buck v. Bell , 274 U.S. 200 (1927). [11] The concept is closely associated with psychology , psychiatry , criminology , and eugenics . However, the term imbecile quickly passed into vernacular usage as a derogatory term. It fell out of professional use in the 20th century in favor of mental retardation . [12] Phrases such as "mental retardation", "mentally retarded", and "retarded" are also subject to the euphemism treadmill : initially used in a medical manner, they gradually took on derogatory connotation .
Using a cholinesterase technique on skin biopsies from the pad of the great toe of affected persons, Dyck et al. (1965) found normal numbers of Meissner corpuscles in a 14-year-old boy with early signs suggestive of the disorder, but no corpuscles in a 37-year-old man and a 28-year-old woman with well-developed disease.
Summary Clinical characteristics. SPTLC1 -related hereditary sensory neuropathy (HSN) is an axonal form of hereditary motor and sensory neuropathy distinguished by prominent early sensory loss and later positive sensory phenomena including dysesthesia and characteristic "lightning" or "shooting" pains. Loss of sensation can lead to painless injuries, which, if unrecognized, result in slow wound healing and subsequent osteomyelitis requiring distal amputations. Motor involvement is present in all advanced cases and can be severe. After age 20 years, the distal wasting and weakness may involve proximal muscles, possibly leading to wheelchair dependency by the seventh or eighth decade. Sensorineural hearing loss is variable. Diagnosis/testing. The diagnosis of SPTLC1 -related HSN is established in a proband with characteristic clinical features and identification of a heterozygous pathogenic variant in SPTLC1 on molecular genetic testing.
Hereditary sensory neuropathy type I (HSN I) is a slowly progressive neurological disorder characterised by prominent predominantly distal sensory loss, autonomic disturbances, autosomal dominant inheritance, and juvenile or adulthood disease onset. Epidemiology The exact prevalence is unknown, but is estimated as very low. Clinical description Disease onset varies between the 2nd and 5th decade of life. The main clinical feature of HSN I is a reduction of sensation sense, mainly distributed around the distal parts of the upper and lower limbs. Variable distal muscle weakness and wasting, and chronic skin ulcers are characteristic.
As mutations in the gene affect the same enzyme as those in the SPTLC1 gene, the molecular basis of the disease is suggested to be the same as that of HSAN IA. [28] HSAN ID [ edit ] HSAN ID is caused by heterozygous missense mutations in the ATL1 gene which encodes atlastin-1 . [29] Atlastin-1 is a member of the dynamin/Mx/guanylate-binding protein superfamily of large GTPases . ... These features were thought to result from the genetic diversity of HSAN I (i.e. the expression of different genes, different alleles of a single gene, or modifying genes) or environmental factors. [40] Molecular genetic studies later confirmed the genetic diversity of the disease. [41] Subtype Gene or locus Mutation (DNA/Amino acid) * Clinical features Age of onset OMIM * IA SPTLC1 399T>G/C133W; 398G>A/C133Y; [12] [13] 431T>A/V144D [12] Predominant loss of pain and temperature sensation; sometimes initial sign with long preservation of vibration sense; burning and lancinating pains; ulcerative mutilations; variable distal motor involvement Adolescence * 162400 IB 3p24-p22 [26] [27] unknown Predominant sensory neuropathy with cough and gastroesophageal reflux ; foot ulcerations (rare) Adulthood 608088 IC SPTLC2 [28] 1075G>A/V359M; 1145G>T/G382V; 1510A>T/I504F Loss of pain and temperature sensation; lancinating pain; ulcerative mutilations; variable distal motor involvement; acro-mutilating complications Adulthood * [42] 613640 ID ALT1 [29] 196G>C/E66Q; 976delG/[V326WfsX8] * Severe distal sensory loss and amyotrophy in lower limbs; trophic skin and nail changes; ulcerative mutilations Adulthood 613708 IE DNMT1 [35] 1484A>G/Y495C; 1470-1472TCC>ATA/D490E-P491Y * Loss of all somatosensory modalities; lancinating pain; ulcerative mutilations; sensorineuronal hearing loss, dementia Adulthood 614116 ^DNA; A: adenine, T: thymine, G: guanine, C: cytosine. ... "Epigenetic modifications and human disease". Nature Biotechnology . 28 (10): 1057–68. doi : 10.1038/nbt.1685 .
A number sign (#) is used with this entry because of evidence that hereditary sensory neuropathy type IF (HSN1F) is caused by heterozygous mutation in the ATL3 gene (609369) on chromosome 11q13. Description Hereditary sensory neuropathy type IF is an autosomal dominant sensory neuropathy affecting the lower limbs. Distal sensory impairment becomes apparent during the second or third decade of life, resulting in painless ulceration of the feet with poor healing, which can progress to osteomyelitis, bone destruction, and amputation. There is no autonomic involvement, spasticity, or cognitive impairment (summary by Kornak et al., 2014). For a discussion of genetic heterogeneity of HSN, see HSAN1A (162400).
A number sign (#) is used with this entry because hereditary sensory and autonomic neuropathy type IC (HSAN1C) is caused by heterozygous mutation in the SPTLC2 gene (605713) gene on chromosome 14q24. For a discussion of genetic heterogeneity of HSAN, see HSAN1A (162400). Clinical Features Rotthier et al. (2010) reported 4 unrelated probands with hereditary sensory neuropathy. Three probands had adult onset (ages 38, 37, and 52 years) of distal sensory loss. The presenting symptoms in these patients included loss of touch and vibration in the feet, dysesthesia and severe panmodal sensory loss in the upper and lower limbs, and distal lower limb sensory loss with ulceration and osteomyelitis necessitating amputation of the right great toe.
A number sign (#) is used with this entry because autosomal dominant hereditary sensory neuropathy type 1D (HSN1D) is caused by heterozygous mutation in the atlastin-1 gene (ATL1; 606439) on chromosome 14q. Description Autosomal dominant hereditary sensory neuropathy type 1D is characterized by adult onset of a distal axonal sensory neuropathy affecting all modalities, often associated with distal ulceration and amputation as well as hyporeflexia, although some patients may show features suggesting upper neuron involvement (summary by Guelly et al., 2011). For a discussion of genetic heterogeneity of HSAN, see HSAN1A (162400). Spastic paraplegia-3A (SPG3A; 182600) is an allelic disorder with a different phenotype. Clinical Features Guelly et al. (2011) reported a 4-generation family with hereditary sensory neuropathy inherited in an autosomal dominant pattern.
Hereditary sensory neuropathy type 1 (HSN1) is a neurological condition characterized by nerve abnormalities in the legs and feet. Many people with this condition have tingling, weakness, and a reduced ability to feel pain and sense hot and cold. Some affected people do not lose sensation, but instead feel shooting pains in their legs and feet. As HSN1 progresses, sensory problems can affect the hands, arms, shoulders, and abdomen. In rare cases, people with this condition develop sensorineural hearing loss.
. ^ " Fast-Food Wrappers Could Increase Miscarriage Risk by 16 Times BY CONOR GAFFEY 4/28/15 AT 11:36 AM, Newsweek . ^ The Lost World of Communism , Peter Molloy, BBC Books, 2009. ^ Medrea, N.; Dumitrescu, I.; Toader, 0.; Tachescu, A. (1984): Toxic abortion in cows due to nitrate-nitrite poisoning. ^ Correlations between the Degree of Heavy Metal Contamination of Fodder and their Accumulation in Organs and Tissues , Adriana Amfim, Violeta Elena Simion, Monica Pârvu, Spiru Haret University, Bucharest, Faculty of Veterinary Medicine, 032091-Bucharest Energeticienilor Blvd, 3, 9-11 Romania, page 3, "The main diseases recorded were: gastroenteritis, maternal toxicosis, lung and liver diseases, toxic abortion, marasmatic syndrome, nephropathy." ^ Tibary, Ahmed.
In the late 1980s, he resided in Omaha , Nebraska and died on September 28, 1992 in Florida, after suffering a stroke before he could deliver a speech to a forum. [12] References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006).
A number sign (#) is used with this entry because of evidence that X-linked lymphoproliferative syndrome-2 (XLP2) is caused by mutation in the gene encoding the X-linked inhibitor of apoptosis (XIAP; 300079) on chromosome Xq25. Description XLP2 is an X-linked primary immune deficiency with symptom onset usually in the first years of life, although later onset may occur. Features are compatible with immune dysregulation and include hemophagocytic lymphohistiocytosis (HLH), often associated with chronic Epstein-Barr virus (EBV) infection, splenomegaly, fever, colitis or inflammatory bowel disease (IBD), and recurrent infections. Laboratory abnormalities are variable, but can include hypogammaglobulinemia, cytopenias, and low levels of a particular subset of T cells known as NKT (or iNKT) cells. Functional studies show increased sensitivity of T cells to apoptosis (activation-induced cell death, AICD), impaired cytokine production, including of TNF-alpha (TNFA; 191160), and general dysregulation of the immune pathway, such as increased levels of IL18 (600953).
X-linked lymphoproliferative syndrome (XLP) is an immune system disorder that occurs almost exclusively in males. People with XLP have an increased risk of infection because their body cannot properly regulate the number of immune system cells (lymphocytes) and blood cells. The symptoms associated with XLP vary from person to person, and even among family members. In most cases, symptoms begin anywhere from 6 months of age to 10 years of age. XLP generally has two subtypes, which are caused by mutations in different genes: XLP1 is mainly characterized by an inappropriate immune response to Epstein-Barr virus (EBV) infection , leading to hemophagocytic lymphohistiocytosis (HLH) or severe mononucleosis ; dysgammaglobulinemia; and lymphoproliferative disease (malignant lymphoma).
Harris et al. (1988) excluded linkage of XLP to 28 X-linked probes. Harris and Docherty (1988) found no particular chromosomal abnormalities in this disorder.
Examples in science [ edit ] William Thomson, 1st Baron Kelvin writing to George FitzGerald on April 9, 1896: [15] "I have not had a moment's peace or happiness in respect to electromagnetic theory since Nov. 28, 1846 (see vol i. p. 80 M.P.P). "All this time I have been liable to fits of ether dipsomania, kept away at intervals only by rigorous abstention from thought on the subject."
The genetic influence is indicated by studies showing that (1) there is a 25 to 50% lifetime risk for alcoholism in sons and brothers of severely alcoholic men; (2) alcohol preference can be selectively bred for in experimental animals; (3) there is a 55% or higher concordance rate in monozygotic twins with only a 28% rate for like-sex dizygotic twins; and (4) half brothers with different fathers and adopted sons of alcoholic men show a rate of alcoholism more like that of the biologic father than that of the foster father.
Prohibition did not work. [21] Treatment of alcoholism may take several forms. [9] Due to medical problems that can occur during withdrawal, alcohol detoxification should be carefully controlled. [9] One common method involves the use of benzodiazepine medications, such as diazepam . [9] These can be either given while admitted to a health care institution or occasionally while a person remains in the community with close supervision. [9] Mental illness or other addictions may complicate treatment. [22] After detoxification, various forms of individual or group therapy or support groups can help keep a person from returning to drinking. [8] [23] One commonly used form of support is the group Alcoholics Anonymous . [24] The medications acamprosate , disulfiram or naltrexone may also be used to help prevent further drinking. [10] The World Health Organization has estimated that as of 2016, there were 380 million people with alcoholism worldwide (5.1% of the population over 15 years of age). [11] [12] As of 2015 in the United States, about 17 million (7%) of adults and 0.7 million (2.8%) of those age 12 to 17 years of age are affected. [13] Alcoholism is most common among males and young adults. [4] Geographically, it is least common in Africa (1.1% of the population) and has the highest rates in Eastern Europe (11%). [4] Alcoholism directly resulted in 139,000 deaths in 2013, up from 112,000 deaths in 1990. [25] A total of 3.3 million deaths (5.9% of all deaths) are believed to be due to alcohol. [13] Alcoholism reduces a person's life expectancy by approximately ten years. [26] Many terms, some insulting and others informal , have been used to refer to people affected by alcoholism; the expressions include tippler , drunkard , dipsomaniac and souse . [27] In 1979, the World Health Organization discouraged the use of "alcoholism" due to its inexact meaning, preferring "alcohol dependence syndrome". [28] Contents 1 Signs and symptoms 1.1 Long-term misuse 2 Alcohol abuse 2.1 Warning signs 2.1.1 Physical 2.1.1.1 Short-term effects 2.1.1.2 Long-term effects 2.1.2 Psychiatric 2.1.3 Social effects 2.2 Alcohol withdrawal 3 Causes 3.1 Availability 3.2 Gender difference 3.3 Genetic variation 4 Diagnosis 4.1 Definition 4.1.1 Alcoholism 4.1.2 DSM and ICD 4.2 Social barriers 4.3 Screening 4.4 Urine and blood tests 5 Prevention 6 Management 6.1 Detoxification 6.2 Psychological 6.3 Moderate drinking 6.4 Medications 7 Disulfiram-like drug 7.1 Dual addictions and dependences 8 Epidemiology 9 Prognosis 10 History 11 Society and culture 12 See also 13 References 14 External links Signs and symptoms Play media Effects of alcohol on the body The risk of alcohol dependence begins at low levels of drinking and increases directly with both the volume of alcohol consumed and a pattern of drinking larger amounts on an occasion , to the point of intoxication, which is sometimes called "binge drinking". ... The World Health Organization uses the term "alcohol dependence syndrome" rather than alcoholism. [28] The concept of "harmful use" (as opposed to "abuse") was introduced in 1992's ICD-10 to minimize underreporting of damage in the absence of dependence. [103] The term "alcoholism" was removed from ICD between ICD-8/ICDA-8 and ICD-9 . [107] ICD-11 Episode of harmful use of alcohol, Dipsomania, Harmful pattern of use of alcohol, or Alcohol dependence Episode of harmful use of alcohol - "A single episode of use of alcohol that has caused damage to a person’s physical or mental health or has resulted in behaviour leading to harm to the health of others ...." [108] Harmful pattern of use of alcohol - "A pattern of alcohol use that has caused damage to a person’s physical or mental health or has resulted in behaviour leading to harm to the health of others ...." [109] Alcohol dependence - "Alcohol dependence is a disorder of regulation of alcohol use arising from repeated or continuous use of alcohol.
Alcohol use disorder is a diagnosis made when an individual has severe problems related to drinking alcohol. Alcohol use disorder can cause major health, social, and economic problems, and can endanger affected individuals and others through behaviors prompted by impaired decision-making and lowered inhibitions, such as aggression, unprotected sex, or driving while intoxicated. Alcohol use disorder is a broad diagnosis that encompasses several commonly used terms describing problems with drinking. It includes alcoholism, also called alcohol addiction, which is a long-lasting (chronic) condition characterized by a powerful, compulsive urge to drink alcohol and the inability to stop drinking after starting. In addition to alcoholism, alcohol use disorder includes alcohol abuse, which involves problem drinking without addiction.