Asplenic patients are particularly susceptible to infection by capnocytophaga canimorsus and should receive a five-day course of amoxicillin/clavulanate ( erythromycin in patients allergic to penicillin). [28] Tick bites - Babesiosis is a rare tickborne infection. ... Quinine (with or without clindamycin ) is usually an effective treatment. [28] Alert warning - People without a working spleen can carry a card, or wear a special bracelet or necklet which says that they do not have a working spleen. ... Archived from the original (PDF) on 2007-09-28 . Retrieved 2006-12-12 . - reprint from Kent and Medway NHS and Social Care Partnership Trust ^ a b Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force (1996). ... Baylor University Medical Center Proceedings. 2015 Jul; 28(3): 340–341 . 28 (3): 340–1. doi : 10.1080/08998280.2015.11929267 .
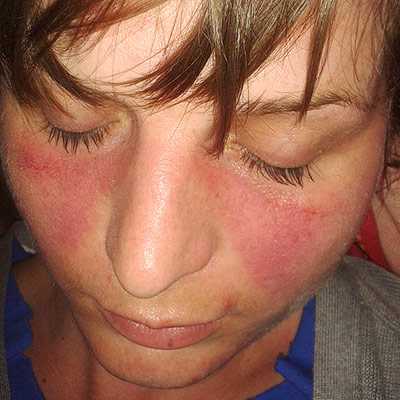
Cold agglutinin disease Specialty Hematology Cold agglutinin disease ( CAD ) is a rare autoimmune disease characterized by the presence of high concentrations of circulating cold sensitive antibodies , usually IgM and autoantibodies that are also active at temperatures below 30 °C (86 °F), [1] directed against red blood cells , causing them to agglutinate and undergo lysis . [2] It is a form of autoimmune hemolytic anemia , specifically one in which antibodies bind red blood cells only at low body temperatures, typically 28–31 °C. When affected people's blood is exposed to cold temperatures (32 °F (0 °C; 273 K) to 50 °F (10 °C; 283 K)), certain proteins that normally attack bacteria (IgM antibodies) attach themselves to red blood cells and bind them together into clumps (agglutination). ... In individuals with cold agglutinin disease, these antibodies are in much higher concentrations (titers over 1000 at 4 °C). At body temperatures of 28–31 °C, such as those encountered during winter months, and occasionally at body temperatures of 37 °C, antibodies (generally IgM ) bind to the polysaccharide region of glycoproteins on the surface of red blood cells (typically the I antigen , i antigen , and Pr antigens ). [10] Binding of antibodies to red blood cells activates the classical pathway of the complement system . ... The auto antibodies responsible for hemagglutination at low temperatures, cold agglutinins (CA), may be found in the sera of healthy subjects as well as in patients with AIHA of the cold reactive types. [34] [32] [25] CA bind to erythrocyte surface antigens at a temperature optimum of 0–4°C. [28] [35] In contrast to polyclonal CA in healthy individuals, monoclonal CA often have a high-thermal amplitude, which contributes to their pathogenicity at temperatures approaching 37°C. [28] [35] [36] [25] Binding of CA causes agglutination of erythrocytes [32] [33] [37] and the antigen–antibody complex induces complement (C) activation and hemolysis. [19] [38] Essential clinical manifestations of primary CAD are hemolytic anemia and cold-induced circulatory symptoms. [32] [33] [39] Exact estimates of the severity of anemia and the frequency of cold-induced symptoms, however, have not been provided until recent years. [27] [32] [33] [40] [25] See also [ edit ] Cold shock response Donath-Landsteiner hemolytic anemia List of hematologic conditions Paroxysmal cold hemoglobinuria Warm antibody autoimmune hemolytic anemia References [ edit ] ^ a b c "Cold agglutinin disease" . ... NORD (National Organization for Rare Disorders) . 2004-10-28. Archived from the original on 2017-02-21 . ... "Immunopathologic and clinical features of hemolytic anemia due to cold agglutinins". Seminars in Hematology . 28 (1): 66–77. ISSN 0037-1963 . PMID 1708169 . ^ Gertz, MA (2006), "Cold agglutinin disease
Pathologic cold agglutinins occur at titers over 1:1000 and react at 28-31 °C and sometimes at 37 °C. Cold agglutinin disease usually results from the production of a specific IgM antibody directed against the I/i antigens (precursors of the ABH and Lewis blood group substances) on red blood cells (RBCs).
Cold agglutinin disease is a type of autoimmune hemolytic anemia (see this term) defined by the presence of cold autoantibodies (autoantibodies which are active at temperatures below 30°C). Epidemiology Cold agglutinin disease represents an estimated 16-32% of AIHA, whose annual incidence is estimated to be between 1/35,000-1/80,000 in North America and Western Europe. Clinical description It occurs more frequently after the age of 55. Cold agglutinin disease manifests as acute or chronic hemolytic anemia, with associated pallor and fatigue. Symptoms during hemolytic ``crises'' may include severe pain in the back and legs, headache, vomiting, diarrhea, dark urine and hepatosplenomegaly. A cold environment or a concurrent infection may trigger or exacerbate the condition, and episodes of acute hemolysis with hemoglobinemia and hemoglobinuria are more common in winter.
Cold agglutinin disease is a rare type of autoimmune hemolytic anemia in which the body's immune system mistakenly attacks and destroys its own red blood cells. When affected people's blood is exposed to cold temperatures (32º to 50º F), certain proteins that normally attack bacteria (IgM antibodies) attach themselves to red blood cells and bind them together into clumps (agglutination). This eventually causes red blood cells to be prematurely destroyed (hemolysis) leading to anemia and other associated signs and symptoms. Cold agglutinin disease can be primary (unknown cause) or secondary, due to an underlying condition such as an infection, another autoimmune disease, or certain cancers. Treatment depends on many factors including the severity of the condition, the signs and symptoms present in each person, and the underlying cause.
Cold autoimmune hemolytic anemia comprises two types of autoimmune hemolytic anemia (AIHA; see this term) defined by the presence of cold autoantibodies (autoantibodies which are active at temperatures below 30°C): cold agglutinin disease (CAD), which is the more common, and paroxysmal cold hemoglobinuria (PCH; see these terms). Clinical description CAD is more common in people over the age of 55 years, while PCH typically presents in young children. Etiology CAD is caused by IgM autoantibodies while PCH is caused by an IgG immunoglobulin.
Health Canada [14] and the US CDC recommend close contacts see their doctor for full evaluation and may require antibiotics; [15] current UK Health Protection Agency guidance is that, for a number of reasons, close contacts should not receive antibiotics unless they are symptomatic but that they should receive information and advice to seek immediate medical attention if they develop symptoms. [13] However, guidance is clearer in the case of mother-baby pairs: both mother and baby should be treated if either develops an invasive GAS infection within the first 28 days following birth [13] (though some evidence suggests that this guidance is not routinely followed in the UK [16] ). ... This was shown by a training camp located in Texas, where a harmful strain of pneumonia complicating measles was caused by a strain of Streptococcus. [28] Existence of streptococci strains was additionally found in World War II. An epidemic of streptococcal infection in the United States Navy during this war indicated that this type of disease was able to exist and spread in formerly unexposed individuals by environments that serological types of group A streptococci preferred. [28] In later years, a positive test result for the presence of group A streptococci was found in 32.1 percent of individuals after throat cultures were carried out in a 20 yearlong (1953/1954-1973/1974) study performed in Nashville, TN. [28] Also, from 1972-1974, recurring GAS illness was observed with a prevalence of 19 percent in school-aged children as well as a prevalence rate of 25 percent in families. [28] The severity of streptococcal infections has decreased over the years, and so has rheumatic fever (a sequelae of GAS) which is indicated by the change in numerous hospitals from containing wards allocated for the sole purpose of treating rheumatic fever to hardly seeing the disease at all. [28] Environmental factors, such as less crowding and the increase of family living space, can account for the reduction in incidence and severity of group A streptococci. [28] With more space for individuals to reside in, it provides the bacteria with less opportunities to spread from person to person.
Since the early 1980s, a resurgence of severe, invasive infections by group A streptococci (GAS; Streptococcus pyogenes) has occurred. The reemergence of streptococcal toxic shock syndrome (STSS) and necrotizing fasciitis (NF) has been reported in several countries. Both are rapidly progressive invasive diseases that are associated with high mortality rate, ranging from 30 to 80% despite prompt antibiotic therapy and debridement. A particular subclone of the M1 serotype (M1T1) has persisted for more than 20 years as the most prevalent strain isolated from these infections. Release of inflammatory cytokines triggered by streptococcal superantigens has a pivotal role in invasive streptococcal disease.
International Craniofacial Institute . Retrieved Oct 28, 2012 . ^ a b c d e f g h i j k l m Clauser L, Galie M. ... NCBI. PMID 20301368 . Retrieved Oct 28, 2012 . ^ "Saethre-Chotzen Syndrome" . ... Johns Hopkins Medicine . Retrieved November 28, 2012 . ^ "Saethre-Chotzen Syndrome" . Boston Children's Hospital . Retrieved November 28, 2012 . ^ "Saethre-Chotzen Syndrome" . Seattle Children's Hospital . Retrieved November 28, 2012 . External links [ edit ] NIH summary on SCS NIH.
Summary Clinical characteristics. Classic Saethre-Chotzen syndrome (SCS) is characterized by coronal synostosis (unilateral or bilateral), facial asymmetry (particularly in individuals with unicoronal synostosis), strabismus, ptosis, and characteristic appearance of the ear (small pinna with a prominent superior and/or inferior crus). Syndactyly of digits two and three of the hand is variably present. Cognitive development is usually normal, although those with a large genomic deletion are at an increased risk for intellectual challenges. Less common manifestations of SCS include other skeletal findings (parietal foramina, vertebral segmentation defects, radioulnar synostosis, maxillary hypoplasia, ocular hypertelorism, hallux valgus, duplicated or curved distal hallux), hypertelorism, palatal anomalies, obstructive sleep apnea, increased intracranial pressure, short stature, and congenital heart malformations. Diagnosis/testing. The diagnosis of SCS is established in a proband with typical clinical findings and the identification of a heterozygous pathogenic variant in TWIST1 by molecular genetic testing. Management. Treatment of manifestations: Ongoing management by an established craniofacial team which may include cranioplasty in the first year of life and midface surgery in childhood as needed for dental malocclusion, swallowing difficulties, and respiratory problems.
Saethre-Chotzen syndrome is a genetic condition characterized by the premature fusion of certain skull bones (craniosynostosis). This early fusion prevents the skull from growing normally and affects the shape and symmetry of the head and face. Other features may include webbing of certain fingers or toes (syndactyly), small or unusually shaped ears, short stature, and abnormalities of the bones in the spine (the vertebrae). The signs and symptoms of Saethre-Chotzen syndrome vary widely, even among affected individuals in the same family. Mutations (variants) in the TWIST1 gene cause most cases of Saethre-Chotzen syndrome.
A syndrome characterized by unilateral or bilateral coronal synostosis, facial asymmetry, ptosis, strabismus and small ears with prominent superior and/or inferior crus, among other less common manifestations. Epidemiology Saethre-Chotzen syndrome (SCS) prevalence ranges from 1/25,000 to 1/50,000 livebirths. Clinical description SCS has a variable spectrum of manifestations. Classic SCS presents at birth with synostosis of coronal (less commonly in conjunction with sagittal, metopic or lambdoid) sutures resulting in abnormal skull shape, facial asymmetry, low frontal hairline, ptosis, strabismus, tear duct stenosis and small ears with prominent crus. Brachydactyly, broad toes, partial cutaneous syndactyly of digits 2 and 3 of the hand, duplicated distal phalanx of the hallux are also often present. Intelligence is normal in most, but mild to severe developmental delay has been reported, primarily in cases with a large genomic deletion.
A number sign (#) is used with this entry because of evidence that the Saethre-Chotzen syndrome (SCS) is caused by heterozygous mutation in the TWIST1 gene (601622) on chromosome 7p21. See also Muenke syndrome (602849), which is caused by a mutation in the FGFR3 gene (P250R; 134934.0014) and has a similar overlapping phenotype to SCS. In addition, at least 1 individual with a phenotype of SCS has been described with a mutation in the FGFR2 gene (176943.0023). Description Saethre-Chotzen syndrome is characterized by craniosynostosis, facial dysmorphism, and hand and foot abnormalities. Coronal synostosis resulting in brachycephaly is the most frequent cranial abnormality observed, and the most common facial features are asymmetry, hypertelorism, and maxillary hypoplasia.
A single set of steps has been identified to be the most likely theory for autoimmune disease onset. [27] Environmental triggers Reduced oral tolerance Gut dysbiosis Enhanced gut permeability Increased immune reactivity Autoimmunity Chemicals can be found within the direct environment or in the form of drugs, including: hydrazines, hair dyes, trichloroethylene, tartrazines, hazardous wastes, and industrial emissions. [28] UV radiation is found to be a possible cause of development of the autoimmune disease dermatomyositis, [29] exposure to pesticides plays a role in rheumatoid arthritis development, [30] and vitamin D has been found to be a key in preventing immune dysfunctions in older populations. [31] Infectious agents are considered T cell activators, a step needed for activation of autoimmune diseases. ... This is combined with various tests, as no single test can identify an autoimmune disease. [28] Antinuclear antibody [ edit ] A test used to identify abnormal proteins, known as antinuclear antibodies, produced when the body attacks its own tissues. [37] [28] It may test positive in several disorders. This test is most useful for diagnosing systemic lupus erythematosus, having a 95% positive test rate. [38] Complete blood count [ edit ] A test taking measurements on maturity levels, count, and size of blood cells. [28] [37] Targeted cells include: red blood cells, white blood cells, hemoglobin, hematocrit, and platelets. ... If complement is found in low levels, this may be an indication of disease. [37] [28] C reactive protein [ edit ] C reactive protein , a protein made in the liver generally increases with inflammation, and may be high in autoimmune disease. [37] [28] Erythrocyte sedimentation rate [ edit ] This test measures the rate at which a patient’s blood cells descend in a test tube. More rapid descents may indicate inflammation, a common symptom of autoimmune disease. [28] [37] If these tests are indicative antibody abnormalities and inflammation, further tests will be conducted to identify the autoimmune disease present. [28] Treatment [ edit ] Treatment depends on the type and severity of the condition.
The extracellular domain of CR1, which has 25 potential N-glycosylation sites, can be divided into 30 short consensus repeats (SCRs), each having 60 to 70 amino acids with sequence homology between SCRs ranging from 60 to 90%. The first 28 SCRs are further arranged into 4 longer regions termed long homologous repeats (LHRs), designated LHR-A, LHR-B, LHR-C, and LHR-D and consisting of 7 SCRs each.
Multiple loose bodies were removed from various joints of 1 sib--18 from the right hip at about age 26, several from the left elbow at age 28, and 30 from the left hip at age 33.
Springer. pp. 76–. ISBN 978-3-319-10750-9 . ^ Mosby (28 April 2016). Mosby's Dictionary of Medicine, Nursing & Health Professions - Elsevieron VitalSource .
Anisomastia, or mammary asymmetry, is a common problem in developing adolescent girls. Stratakis et al. (2000) evaluated a 22-year-old female patient who had severe anisomastia (which had been repaired by surgery) associated with moderate to severe mental retardation, a stocky body habitus with mild obesity, dysmorphic facies (prominent, upslanting palpebral fissures, beaked nose, and a prominent philtrum), webbed neck, low hairline, and severe bilateral clinodactyly of the third, fourth, and fifth fingers with acral (but not large joint) flexion contractures. A peripheral blood high-resolution karyotype revealed additional chromosomal material within the long arm of chromosome 16. Densitometric analysis of amplified polymorphic sequence-tagged sites (STSs) mapping to 16q suggested that the duplication is defined by the noninvolved markers D16S419 (16q12-cen, 66 cM from 16p terminus) and D16S421 (16q13-q21, 84.4 cM), encompassing a maximum of 18.4 cM of genetic distance. The STS analysis showed that the duplication was on the maternally derived chromosome 16, resulting in 2 maternal (and 1 paternal) copies of that region of chromosome 16.
"Transitional cell carcinoma of the ovary: a morphologic study of 100 cases with emphasis on differential diagnosis". Am J Surg Pathol . 28 (4): 453–63. doi : 10.1097/00000478-200404000-00004 .
The same study found that the chance of having coprolalia increased linearly with the number of comorbid conditions: patients with four or five other conditions—in addition to tics—were four to six times more likely to have coprolalia than persons with only Tourette's. [17] One study of a general pediatric practice found an 8% rate of coprolalia in children with Tourette syndrome, while another study found 60% in a tertiary referral center (where typically more severe cases are referred). [18] A more recent Brazilian study of 44 patients with Tourette syndrome found a 14% rate of coprolalia; [19] a Costa Rican study of 85 subjects found 20% had coprolalia; [20] a Chilean study of 70 patients found an 8.5% rate of coprolalia; [21] older studies in Japan reported a 4% incidence of coprolalia; [22] and a still older clinical study in Brazil found 28% of 32 patients had coprolalia. [23] Considering the methodological issues affecting all of these reports, the consensus of the Tourette Syndrome Association is that the actual number is below 15 percent. ... This does not prevent the vocalizations, but the partial paralysis that results helps to control the volume of any outbursts. [24] [25] [26] Botox injections result in more generalized relief of tics than the vocal relief expected. [27] Society and culture [ edit ] The entertainment industry often depicts those with Tourette syndrome as being social misfits whose only tic is coprolalia, which has furthered stigmatization and the public's misunderstanding of those with Tourette's. [28] [29] [30] The coprolalic symptoms of Tourette's are also fodder for radio and television talk shows. [31] See also [ edit ] List of language disorders References [ edit ] ^ Coprolalia. ... PMID 15721825 . ^ Schapiro NA (2002). " ' Dude, you don't have Tourette's:' Tourette's syndrome, beyond the tics" . Pediatr Nurs . 28 (3): 243–6, 249–53. PMID 12087644 . ^ "Linguistics 210 Semantics" (PDF) . ... Lesch-Nyhan Syndrome. eMedicine.com (August 29, 2006). Accessed 28 October 2006. ^ Tourette Syndrome FAQ.
The median age at onset of neurologic dysfunction was 13 years (range, 5-28 years), and all had distal muscle atrophy and weakness affecting the lower limbs, although the severity was variable; older individuals had greater impairment. ... INHERITANCE - Autosomal dominant HEAD & NECK Ears - Deafness, sensorineural (in some patients) GENITOURINARY Kidneys - Focal segmental glomerulosclerosis - Proteinuria - End-stage renal disease (in some) SKELETAL Hands - Claw hands Feet - Pes cavus - Hammertoes MUSCLE, SOFT TISSUES - Amyotrophy, distal, upper and lower limbs NEUROLOGIC Peripheral Nervous System - Distal limb muscle weakness due to peripheral neuropathy (lower and sometimes upper limbs are affected) - Distal limb muscle atrophy due to peripheral neuropathy - 'Steppage' gait - Foot drop - Hyporeflexia - Areflexia - Distal sensory impairment - Sural nerve biopsy shows axonal loss - Onion bulb formation - Low to normal range of motor nerve conduction velocities (23 to 45 m/sec) ('intermediate' CMT) MISCELLANEOUS - Median onset of proteinuria is 18 years (range 10 to 21) - Median onset of neurologic symptoms is 13 years (range 5 to 28) - Progressive disorder regarding both neurologic and renal symptoms MOLECULAR BASIS - Caused by mutation in the inverted formin 2 gene (INF2, 610982.0006 ) ▲ Close
A rare hereditary motor and sensory neuropathy disorder characterized by the typical CMT phenotype (slowly progressive distal muscle weakness and atrophy in upper and lower limbs, distal sensory loss in extremities, reduced or absent deep tendon reflexes and foot deformities) associated with focal segmental glomerulosclerosis (manifesting with proteinuria and progression to end-stage renal disease). Mild or moderate sensorineural hearing loss may also be associated. Nerve biopsy reveals both axonal and demyelinating changes and nerve conduction velocities vary from the demyelinating to axonal range (typically between 25-50m/sec).
New York: St. Martin's Griffin . pp. 27–28. ISBN 978-0805083392 . ^ Mineka, Susan; Keir, Richard; Price, Veda (1980). ... S.; Tomaz, C.; Tran, A. H.; Ono, T.; Nishijo, H. (28 October 2013). "Pulvinar neurons reveal neurobiological evidence of past selection for rapid detection of snakes" .
PMID 15738120 . ^ Moss, Alex (2020-08-28). "Covid-19 smell loss 'made meat taste like petrol ' " . BBC News . Retrieved 2020-08-28 . ^ Emmett, EA (1976). "Parosmia and hyposmia induced by solvent exposure" .