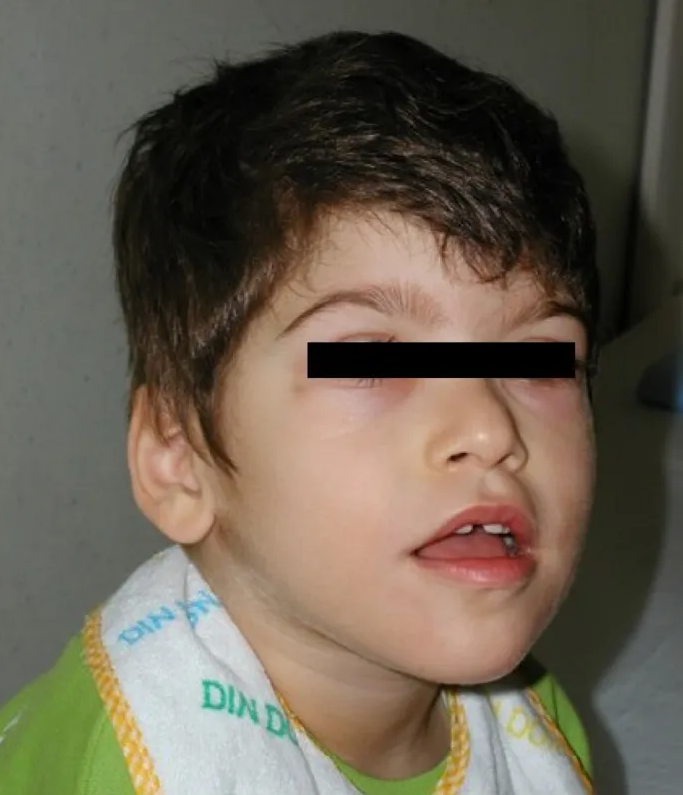
Clinical description Wolf-Hirschhorn syndrome (WHS) occurs more frequently in females than in males (2:1). ... Differential diagnosis Differential diagnosis includes many syndromes displaying growth failure, intellectual disability and/or facial dysmorphism such as Seckel, CHARGE, Smith-Lemli-Opitz, Opitz G/BBB, Williams, Rett, Angelman and Smith-Magenis syndromes (see these terms).
Wolf-Hirschhorn syndrome is a condition that affects many parts of the body. ... Wolf-Hirschhorn syndrome can also cause abnormalities of the eyes, heart, genitourinary tract, and brain. A condition called Pitt-Rogers-Danks syndrome has features that overlap with those of Wolf-Hirschhorn syndrome. ... Frequency The prevalence of Wolf-Hirschhorn syndrome is estimated to be 1 in 50,000 births. ... For unknown reasons, Wolf-Hirschhorn syndrome occurs in about twice as many females as males.
A number sign (#) is used with this entry because Wolf-Hirschhorn syndrome (WHS) is a contiguous gene deletion syndrome associated with a hemizygous deletion of chromosome 4p16.3. ... Lizcano-Gil et al. (1995) described a similar case of what was then called the 'Pitt-Rogers-Danks syndrome (PRDS)' or 'Pitt syndrome,' with the additional feature of optic atrophy. ... Haplotype analysis and investigation with microsatellite and EST markers suggested a disease locus in a region of Xp22, but no evidence for deletion indicative of a contiguous gene deletion syndrome was found. In a clinical and genetic reevaluation of the 2 living affected sibs in this family, Wieland et al. (2014) concluded that the disorder, previously called Wittwer syndrome, is a variant of Wolf-Hirschhorn syndrome (see CYTOGENETICS). ... Battaglia and Carey (1998) also argued that the Pitt-Rogers-Danks syndrome is essentially the same as Wolf-Hirschhorn syndrome, i.e., a 4p deletion syndrome. ... Battaglia et al. (1999) evaluated 15 patients with the 4p- syndrome (12 females, 3 males) in 3 centers.
A number sign (#) is used with this entry because autosomal recessive hyper-IgE recurrent infection syndrome-2 (HIES2) is caused by homozygous or compound heterozygous mutation in the DOCK8 gene (611432) on chromosome 9p24. Description Autosomal dominant hyper-IgE recurrent infection syndrome-1 (HIES1; 147060) is a primary immunodeficiency disorder characterized by recurrent Staphylococcus aureus skin abscesses, increased serum IgE, and abnormalities of the connective tissue, skeleton, and dentition (Buckley et al., 1972; Grimbacher et al., 1999). ... For a discussion of genetic heterogeneity of hyper-IgE recurrent infection syndrome, see 147060. Clinical Features Renner et al. (2004) reported 13 patients from 6 consanguineous families with hyper-IgE syndrome and recurrent infections. ... Molecular Genetics In 11 affected individuals from 8 unrelated families with autosomal recessive hyper-IgE recurrent infection syndrome, Zhang et al. (2009) identified homozygosity or compound heterozygosity for deletions or mutations in the DOCK8 gene (see, e.g., 611432.0001-611432.0005). ... INHERITANCE - Autosomal recessive RESPIRATORY - Recurrent sinopulmonary infections Airways - Asthma SKIN, NAILS, & HAIR Skin - Eczema, severe - Atopic dermatitis - Skin abscesses, recurrent NEUROLOGIC Central Nervous System - Increased neurologic sequelae of infections (rare) - Hemiplegia (rare) - Ischemic infarction (rare) - Subarachnoid hemorrhage (rare) IMMUNOLOGY - Recurrent bacterial infections - Recurrent viral infections - Recurrent fungal infections - Impaired T cell immunity - Impaired T cell proliferation and activation - Food allergies - Environmental allergies NEOPLASIA - Increased susceptibility to carcinomas, especially cancers related to cutaneous viral infections LABORATORY ABNORMALITIES - Increased serum IgE - Eosinophilia - Decreased T cells - Decreased B cells - Decreased natural killer cells - Decreased serum IgM MISCELLANEOUS - Onset in infancy - Early death may occur due to infection - Distinct disorder from autosomal dominant hyper IgE syndrome ( 147060 ) MOLECULAR BASIS - Caused by mutation in the dedicator of cytokinesis 8 gene (DOCK8, 611432.0001 ) ▲ Close
DOCK8 immunodeficiency syndrome is a disorder of the immune system. ... It is unclear why people with DOCK8 immunodeficiency syndrome have such high levels of this protein. ... DOCK8 immunodeficiency syndrome is also commonly called autosomal recessive hyper-IgE syndrome. ... Frequency DOCK8 immunodeficiency syndrome is a rare disorder whose prevalence is unknown. Causes DOCK8 immunodeficiency syndrome is caused by mutations in the DOCK8 gene.
Autosomal recessive hyper IgE syndrome (AR-HIES) is a very rare primary immunodeficiency syndrome characterized by highly elevated blood levels of immunoglobulin E (IgE) , recurrent staphylococcal skin abscesses , and recurrent pneumonia . The same features are also seen in the more frequent autosomal dominant HIES syndrome . AR-HIES accounts for only a small minority of HIES cases, with about 130 affected families reported so far.
The neurologic, vasculitic and autoimmune symptoms associated with autosomal dominant hyper IgE syndrome due to mutations in STAT3 (see this term), are not observed.
DOCK8 deficiency Other names Combined immunodeficiency due to dedicator of cytokinesis 8 protein deficiency, CID due to DOCK8 deficiency DOCKS deficiency is autosomal recessive DOCK8 deficiency , also called DOCK8 immunodeficiency syndrome , is the autosomal recessive form of hyperimmunoglobulin E syndrome , a genetic disorder characterized by elevated immunoglobulin E levels, eosinophilia , and recurrent infections with staphylococcus and viruses. ... It can be distinguished from the similar X-linked Wiskott–Aldrich syndrome by the presence of thrombocytopenia and the consequent bloody diarrhea , as well as its pattern of inheritance . WHIM syndrome , caused by a mutation in CXCR4 , is associated with similar chronic cutaneous viral infections. [4] Treatment [ edit ] Treatment in DOCK8 deficiency focuses on preventing and treating infections. ... Retrieved 2015-07-26 . ^ a b c d e f g h i Szczawinska-Poplonyk, Aleksandra; Kycler, Zdzislawa; Pietrucha, Barbara; Heropolitanska-Pliszka, Edyta; Breborowicz, Anna; Gerreth, Karolina (2011-01-01). "The hyperimmunoglobulin E syndrome--clinical manifestation diversity in primary immune deficiency" . ... "Genetic, clinical, and laboratory markers for DOCK8 immunodeficiency syndrome" . Disease Markers . 29 (3–4): 131–139. doi : 10.3233/DMA-2010-0737 .
Disease FG syndrome Other names Opitz–Kaveggia syndrome, FGS1 Specialty Medical genetics Usual onset Birth Duration Lifelong Risk factors Family history (genetics) FG syndrome ( FGS ) is a rare genetic syndrome caused by one or more recessive genes located on the X chromosome and causing physical anomalies and developmental delays. ... History [ edit ] Kim Peek (1951–2009) probably had FG syndrome. The name of the syndrome comes from the initials of the surnames of two sisters, who had five sons with the syndrome. ... See also [ edit ] Lujan–Fryns syndrome References [ edit ] ^ "Faq Pages" . FG Syndrome Family Alliance . fgsyndrome.org . Retrieved 20 September 2016 . ^ a b Opitz JM, Kaveggia EG (1974). "Studies of malformation syndromes of man XXXIII: the FG syndrome.
A number sign (#) is used with this entry because of evidence that the Opitz-Kaveggia syndrome, also known as FG syndrome-1 (FGS1), is caused by mutation in the MED12 gene (300188) on chromosome Xq13. ... Genetic Heterogeneity of FG Syndrome Other forms of 'FG syndrome' were characterized due to the similar clinical features observed by Opitz and Kaveggia (1974). ... None of the affected males had all features of the FG syndrome and none had macrocephaly. Fetal finger pads are typical of this disorder and of the Kabuki make-up syndrome (147920). ... The boys with FG syndrome had socially oriented, attention-seeking behaviors similar to those of boys with Williams syndrome. These studies confirmed the previous descriptions of a typical personality in FG syndrome. Rauch et al. (1998) considered the report of Chrzanowska et al. (1998), detailing a 'new' branchial syndrome that included foramina parietalia permagna, to represent a case of the FG syndrome.
A rare X-linked syndromic intellectual disability characterized by developmental delay and intellectual disability, early hypotonia, constipation, feeding problems, imperforate anus, characteristic behavior (affable, eager to please), and dysmorphic craniofacial features (such as relative macrocephaly, prominent forehead with frontal hair upsweep, hypertelorism, downslanting palpebral fissures, and open mouth).
FG syndrome (FGS) is a genetic condition that affects many parts of the body and occurs almost exclusively in males. "FG" represents the surname initials of the first individuals diagnosed with the disorder. People with FG syndrome frequently have intellectual disability ranging from mild to severe, hypotonia, constipation and/or anal anomalies, a distinctive facial appearance, broad thumbs and great toes, a large head compared to body size (relative macrocephaly), and abnormalities of the corpus callosum. ... Researchers have identified five regions of the X chromosome that are linked to FG syndrome in affected families. Mutations in the MED12 gene appears to be the most common cause of this disorder, leading to FG syndrome 1. Other genes involved with FG syndrome include FLNA (FGS2), CASK (FGS4), UPF3B (FGS6), and BRWD3 (FGS7).
This syndrome is due to mutations in the RNU4ATAC gene. ... Silver–Russell dwarfism (Russell-Silver Syndrome) 180860 The final height of those with Russell–Silver syndrome often exceeds the height of others with primordial dwarfism, and they tend to have dysmorphic features. ... Their heads may appear to be triangular shaped and large for their small body size. Meier–Gorlin syndrome 224690 Individuals with Meier-Gorlin syndrome often have small ears and no kneecaps. They are also found to have curved clavicles , narrow ribs, and elbow dislocation. Like Russell–Silver syndrome, they usually exceed the height of those with Seckel syndrome and ODPDI and II. ... Retrieved 2012-02-28 . [ dead link ] ^ "Smallest Siblings In the World Bridgette and Brad Jordan" Retrieved on 10 February 2019. v t e Growth and height disorder due to endocrine malfunction Dwarfism Primordial dwarfism Laron syndrome Psychosocial Ateliosis Gigantism v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome
Isolated growth hormone deficiency is a condition caused by a severe shortage or absence of growth hormone. Growth hormone is a protein that is necessary for the normal growth of the body's bones and tissues. Because they do not have enough of this hormone, people with isolated growth hormone deficiency commonly experience a failure to grow at the expected rate and have unusually short stature. This condition is usually apparent by early childhood. There are four types of isolated growth hormone deficiency differentiated by the severity of the condition, the gene involved, and the inheritance pattern. Isolated growth hormone deficiency type IA is caused by an absence of growth hormone and is the most severe of all the types.
A number sign (#) is used with this entry because of evidence that isolated growth hormone deficiency type V (IGHD5) is caused by compound heterozygous mutation in the RNPC3 gene (618016) on chromosome 1p21. One such family has been reported. For general phenotypic information and a discussion of genetic heterogeneity of IGHD, see 262400. Description Isolated growth hormone deficiency type V is characterized by severe postnatal growth failure, delayed bone age without bone dysplasia, and hypoplasia of the anterior pituitary (Argente et al., 2014). Clinical Features Argente et al. (2014) reported 3 sisters, born with normal length to normal-statured nonconsanguineous parents, who developed severe postnatal proportionate growth retardation. They showed features typical of growth hormone (GH; see 139250) deficiency, including marked short stature, frontal bossing, cherubic face, mild microcephaly, delayed bone maturation without bone dysplasia, and otherwise normal development.
Oculocerebral hypopigmentation syndrome, Cross type is a rare congenital syndrome characterized by cutaneous and ocular hypopigmentation, various ocular anomalies (e.g. corneal and lens opacity, spastic ectropium, and/or nystagmus), growth deficiency, intellectual deficit and other progressive neurologic anomalies such as spastic tetraplegia, hyperreflexia, and/or athetoid movements.
Clinical Features Cross et al. (1967) described a family in which 4 sibs (2 male, 2 female) had cutaneous hypopigmentation, severe ocular anomalies, and cerebral defect manifested by spasticity, mental and physical retardation, and athetoid movements. The syndrome has often been referred to as Cross syndrome, but it might also be called Kramer syndrome after the family in which it was observed. ... White et al. (1993) described this disorder in a 20-year-old man who also had Bartter syndrome (see 241200), apparently as a coincidental association.
Clinical Features Margolis (1962) described a 'new' X-linked syndrome--deaf-mutism (profound deafness) and total albinism. Also from Israel, Ziprkowski et al. (1962) described an X-linked syndrome consisting of congenital deafness and partial albinism (without ocular albinism). ... This raised the possibility that this syndrome represents only 1 clinical aspect of another allelic X-linked disorder. Zlotogora (1995) proposed Waardenburg syndrome type II (see 193510) as its possible allelic syndrome. ... He suggested that the albinism-deafness syndrome is an X-linked dominant form of Waardenburg syndrome 2, defining WS2 on phenotypic grounds characterized particularly by the absence of dystopia canthorum.
A rare disorder characterised by congenital nerve deafness and piebaldness with no ocular albinism. It has been described in one large pedigree. Transmission is X-linked with affected males presenting with profound sensorineural deafness and severe pigmentary abnormalities of the skin, and carrier females presenting with variable hearing impairment without any pigmentary changes. The causative gene has been mapped to Xq26.3-q27.1.
Researchers sometimes refer to 1q21.1 microdeletion as the recurrent distal 1.35-Mb deletion to distinguish it from the genetic change that causes thrombocytopenia-absent radius syndrome (TAR syndrome). TAR syndrome results from the deletion of a different, smaller DNA segment in the chromosome 1q21.1 region near the area where the 1.35-Mb deletion occurs. The chromosomal change related to TAR syndrome is often called the 200-kb deletion.
1q21.1 microdeletion syndrome is a chromosome abnormality where a segment of genetic material on the long arm (or q arm) of chromosome 1 at position 21.1 is missing (or deleted). ... Psychiatric and behavioral features can include autism spectrum disorders , anxiety and mood disorders, schizophrenia , attention deficit hyperactivity disorder and sleep disorders. This syndrome is caused by a deletion in a specific region of 1q21.1, which is distinct from the deletion region that causes TAR syndrome .
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome (chr1: 145.0-146.4 Mb, NCBI36). See 274000 for another contiguous gene deletion syndrome, thrombocytopenia-absent radius (TAR) syndrome, that maps to a nonoverlapping region of chromosome 1q21.1.
1q21.1 microdeletion syndrome is a newly described recurrent deletion syndrome with variable clinical manifestations but without the clinical picture of thrombocytopenia - absent radius (TAR) syndrome. ... Autism spectrum disorders, schizophrenia or attention deficit hyperactivity disorder have been noted occasionally. Etiology This syndrome is caused by a recurrent 1.35Mb deletion in the distal 1q21.1 region distinct from the deletion region implicated in TAR syndrome (see this term).
A congenital, X-linked, clinical subtype of L1 syndrome, characterized by spastic paraplegia, mild to moderate intellectual disability and normal brain morphology. This subtype represents the milder end of the L1 syndrome spectrum.
A rare, congenital X-linked developmental disorder characterized by hydrocephalus of varying degrees of severity, intellectual deficit, spasticity of the legs, and adducted thumbs. The syndrome represents a spectrum of disorders including: X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS), MASA syndrome, X-linked complicated hereditary spastic paraplegia type 1, and X-linked complicated corpus callosum agenesis. Epidemiology L1 syndrome primarily affects males. HSAS is the most common genetic form of congenital hydrocephalus, with a prevalence of approximately 1/30,000. ... Adducted thumbs are a characteristic feature of the syndrome, present in about 50% of cases. ... A small number of patients (< 20) have been reported to have a combination of L1 syndrome and Hirschsprung disease. Female carriers may have minor features such as adducted thumbs or mild intellectual deficit but they rarely have the severe manifestations of the syndrome. Etiology L1 syndrome is caused by mutations in the L1CAM gene (Xq28) encoding the L1 cell adhesion molecule that is expressed mainly in the developing nervous system.
A X-linked, clinical subtype of L1 syndrome, characterized by mild to moderate intellectual disability, delayed development of speech, hypotonia progressing to spasticity or spastic paraplegia, adducted thumbs, and mild to moderate distension of the cerebral ventricles.
L1 syndrome is a mild to severe congenital disorder with hydrocephalus of varying degrees of severity, intellectual disability, spasticity of the legs, and adducted thumbs. It includes several conditions, some more severe than others: X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS) - the most severe of all; MASA syndrome (intellectual disability, aphasia (delayed speech), spastic paraplegia (shuffling gait), adducted thumbs); SPG1 (X-linked complicated hereditary spastic paraplegia type 1) X-linked complicated corpus callosum agenesis .
Diagnosis Suggestive Findings L1 syndrome involves a phenotypic spectrum ranging from severe to mild. L1 syndrome should be suspected in individuals with any of the following clinical phenotypes or neuroimaging findings and supportive family history. ... Clinical Characteristics Clinical Description L1 syndrome is seen almost exclusively in males. ... Rarely do females manifest the complete L1 syndrome phenotype. Severe hydrocephalus has been reported in a heterozygous female [Kaepernick et al 1994, Vos et al 2010]. ... Nonsyndromic congenital hydrocephalus. Individuals with L1 syndrome and hydrocephalus do not have major additional physical anomalies.
L1 syndrome Other names L1CAM syndrome, CRASH syndrome, Corpus callosum hypoplasia-retardation-adducted thumbs-spasticity-hydrocephalus syndrome Specialty Pediatrics , neurology , medical genetics Usual onset Neonatal Duration Lifelong Risk factors Family history Diagnostic method Genetic testing Treatment Supportive Prognosis Varies depending on specific disorder Frequency Unknown; HSAS 1 per 30,000 male live births L1 syndrome is a group of mild to severe X-linked recessive disorders that share a common genetic basis. The spectrum of L1 syndrome disorders includes X-linked complicated corpus callosum agenesis, spastic paraplegia type 1, MASA syndrome , and X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS). [1] [2] It is also called L1CAM syndrome (for the disorder's causative gene ) and CRASH syndrome, an acronym for its primary clinical features: corpus callosum hypoplasia, retardation ( intellectual disability ), adducted thumbs, spasticity , and hydrocephalus . [2] L1 syndrome can be caused by different variants in L1CAM , [3] the gene that provides the information that allows the body to produce L1 cell adhesion molecule (sometimes called the L1 protein). [3] The L1 cell adhesion molecule is a surface protein found on the surface of all neurons . [4] It allows neurons to bind to one another and create synapses (connections where information is passed on from the axons of one neuron to the dendrites and cell body of another). [4] [5] As a result, L1 cell adhesion molecule is essential for the structural development of the brain and contributes to the ability to think, move, and develop memories. [4] The type and severity of L1CAM variant causing L1 syndrome in a particular person is directly related to the severity of symptoms and functional impairment that they experience. [6] [7] There is no cure for L1 syndrome, and prognosis is often poor. [8] [9] Life expectancy for people with L1 syndrome can vary dramatically depending on the severity of the condition, with some dying shortly after birth and others reaching adulthood. [2] Treatment for people with L1 syndrome is supportive and aims to improve quality of life and minimize functional impairment. [10] [3] Contents 1 Signs and symptoms 1.1 Societal Implications Based on Symptoms 1.2 X-linked hydrocephalus with stenosis of the aqueduct of Sylvius 1.3 MASA syndrome 2 Diagnosis 3 Management 4 References 5 External links Signs and symptoms [ edit ] L1 syndrome presents as a spectrum ranging from mild to severe features. [3] There is a genotype -phenotype correlation across the L1 spectrum, meaning that the specific genetic variant causing an L1-spectrum disorder in a patient determines the severity of the L1 syndrome in that patient. [6] Patients with truncating (loss-of-function) variants in L1CAM , which prevent the full synthesis of L1 (protein) experience more severe features than patients with missense variants in L1CAM , which may result in an abnormal protein but do not prevent its synthesis. [6] Illustrating this difference in L1 syndrome severity, up to 50% of infants born with L1 syndrome caused by a truncating mutation will die before the age of 3 years despite provision of best available medical treatment. [6] In comparison, roughly 10% of infants born with L1 syndrome caused by a missense mutation will die before the age of 3 years. [6] Despite its presentation on a continuous spectrum, L1 syndrome is loosely divided into four discrete phenotypes. [11] [2] Societal Implications Based on Symptoms [ edit ] People diagnosed with L1 syndrome often experience issues with respect to societal roles and interactions due to the severe physical and mental disabilities associated with the disorder. ... "Genotype-phenotype correlations in L1 syndrome: a guide for genetic counselling and mutation analysis" (PDF) . ... "L1CAM whole gene deletion in a child with L1 syndrome". American Journal of Medical Genetics Part A . 164 (6): 1555–1558. doi : 10.1002/ajmg.a.36474 . ... External links [ edit ] Classification D ICD - 10 : Q04.8 External resources GeneReviews : L1 Syndrome Orphanet : 275543 v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia
A number sign (#) is used with this entry because MASA syndrome, also known as spastic paraplegia-1 (SPG1), is caused by mutation in the gene encoding the L1 cell adhesion molecule (L1CAM; 308840). ... Winter et al. (1989) reviewed the similarities of the syndromes reported by Bianchine and Lewis (1974), Gareis and Mason (1984), Yeatman (1984), and Kenwrick et al. (1986) and suggested that they were all compatible with the MASA syndrome. ... Rietschel et al. (1991) emphasized clinical variability of the MASA syndrome and X-linked complicated and pure hereditary spastic paraplegias and noted the overlap of the syndromes. ... However, they suggested that the MASA syndrome is separate from X-linked mental retardation with bilateral clasped thumbs. ... Because of the same linkage relationships on Xq28 in their patients with MASA syndrome and because of the cerebral abnormalities reported in these patients and the occurrence of congenital hydrocephalus in a cousin, Schrander-Stumpel et al. (1990) suggested that X-linked aqueductal stenosis (HSAS; 307000) and the MASA syndrome are allelic disorders.
Marshall syndrome Other names Deafness, myopia, cataract, saddle nose-Marshall type [1] Marshall syndrome and Stickler syndrome is inherited in an autosomal dominant pattern. ... Cataracts also occur more frequently and detached retina less frequently than in Stickler syndrome. Myopia also is the most common problem with the eyes in Stickler syndrome . ... There also may be changes in the bones that show up on X-ray but generally are not a problem. [ citation needed ] Orofacial Structure [ edit ] Main article: Pierre Robin syndrome The most severe problem associated with Stickler syndrome is Pierre Robin syndrome . ... A conductive loss due to otitis can magnify an existing sensorineural loss and is a frequent problem for children with Stickler or Marshall Syndrome. [ citation needed ] Genetics [ edit ] Stickler syndrome and Marshall syndrome have an autosomal dominant pattern of inheritance. ... There are no tests for Stickler syndrome or Marshall syndrome. Some families with Stickler syndrome have been shown to have mutations in the Type II collagen gene on chromosome 1 .
The affected individuals had ectodermal dysplasia and ocular hypertelorism, features not shared by Stickler syndrome (see 108300), Wagner syndrome (143200), or Weissenbacher-Zweymuller syndrome (WZS; 184840), all of which are conditions often confused with Marshall syndrome. ... Winter et al. (1983) described families demonstrating nosologic overlap among Weissenbacher-Zweymuller syndrome, Stickler syndrome, and Marshall syndrome. ... The results demonstrated allelism of Marshall syndrome with a subset of Stickler syndrome families associated with COL11A1 mutations. ... By screening patients with Stickler syndrome, Stickler-like syndrome, or Marshall syndrome, Annunen et al. (1999) identified 23 novel mutations of the COL11A1 gene. ... Ala-Kokko and Shanske (2009) stated that this was the first report of mosaicism in Marshall syndrome. Nomenclature Note that the designation 'Marshall syndrome' has sometimes been used for the Marshall-Smith syndrome; see 602535.
A malformation syndrome that is characterized by facial dysmorphism, severe hypoplasia of the nasal bones and frontal sinuses, ocular involvement, early-onset hearing loss, skeletal and anhidrotic ectodermal anomalies and short stature with spondyloepiphyseal dysplasia and early-onset osteoarthritis.
Periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis (PFAPA) is characterized by short episodes of illness that occur at regular time periods. The syndrome usually occurs in children younger than five years; but it has also been reported in adults.
A condition similar to Stickler syndrome, called Marshall syndrome, is characterized by a distinctive facial appearance, eye abnormalities, hearing loss, and early-onset arthritis. Marshall syndrome can also include short stature. Some researchers have classified Marshall syndrome as a variant of Stickler syndrome, while others consider it to be a separate disorder. Frequency Stickler syndrome affects an estimated 1 in 7,500 to 9,000 newborns. ... These cases occur in people with no history of Stickler syndrome in their family. Marshall syndrome also typically has an autosomal dominant pattern of inheritance.
Marshall syndrome is an inherited condition characterized by a distinctive facial appearance, eye abnormalities, hearing loss, and early-onset arthritis. Those with Marshall syndrome can also have short stature. Some researchers have argued that Marshall syndrome represents a variant form of Stickler syndrome ; but this remains controversial. Marshall syndrome is caused by mutations in the COL11A1 gene and is inherited in an autosomal dominant fashion.
Summary Clinical characteristics. Cockayne syndrome (referred to as CS in this GeneReview ) spans a continuous phenotypic spectrum that includes: CS type I, the "classic" or "moderate" form; CS type II, a more severe form with symptoms present at birth; this form overlaps with cerebrooculofacioskeletal (COFS) syndrome; CS type III, a milder and later-onset form; COFS syndrome, a fetal form of CS. ... Clinical Characteristics Clinical Description Before the molecular genetics of Cockayne syndrome was understood, it was thought to have a single, discrete phenotype: classic Cockayne syndrome. ... Major features only become apparent after age two years. COFS Syndrome COFS syndrome is the most severe subtype of the CS spectrum and can be identified during fetal life. ... ? Hallerman-Streiff syndrome (OMIM 234100) ? ? Rubinstein-Taybi syndrome CREBBP EP300 AD Silver-Russell syndrome See footnote 2 Seckel syndrome (OMIM PS210600) ~9 genes 3 AR Wiedemann-Rautenstrauch syndrome (OMIM 264090) POLR3A AR Congenital infections (e.g., rubella or toxoplasmosis) N/A N/A Calcifications on brain imaging White matter hypomyelination Distinctive physical appearance Disorders of calcium & phosphate metabolism Xeroderma pigmentosum 9 genes 4 AR Prominent photosensitivity &/or thinning of skin & hair White matter hypomyelination Brain calcifications Ataxia Spasticity Neurodegenerative features Bloom syndrome BLM AR Hutchinson-Gilford progeria syndrome LMNA AD Werner syndrome WRN AR Rothmund-Thompson syndrome RECQL4 AR Mitochondrial disorders See footnote 5 AD AR Mat Early presence of pigmentary retinopathy Brain calcifications Neurodegeneration Skin photosensitivity Distinctive physical appearance ? = unknown; AD = autosomal dominant; AR = autosomal recessive; COFS = cerebrooculofacioskeletal syndrome; CS = Cockayne syndrome; DiffDx = differential diagnosis; Mat = maternal; MOI = mode of inheritance; XL = X-linked 1.
A number sign (#) is used with this entry because of evidence that cerebrooculofacioskeletal syndrome-3 (COFS3) is caused by homozygous mutation in the ERCC5 gene (133530) on chromosome 13q33. Biallelic mutations in the ERCC5 gene can also cause Xeroderma pigmentosum, group G and/or Cockayne syndrome (278780). Description Cerebrooculofacioskeletal syndrome is a severe, progressive neurologic disorder characterized by prenatal onset of arthrogryposis, microcephaly, and growth failure. ... COFS represents the severe end of the spectrum of disorders caused by mutations in nucleotide excision repair (NER) genes, with Cockayne syndrome and xeroderma pigmentosum being milder NER-related phenotypes (summary by Drury et al., 2014). For a phenotypic description and a discussion of genetic heterogeneity of cerebrooculofacioskeletal syndrome, see COFS1 (214150). Clinical Features Hamel et al. (1996) and Nouspikel et al. (1997) studied a male who was born to healthy first-cousin Moroccan parents and had extremely severe early-onset Cockayne syndrome leading to death at 7 months of age. Graham et al. (2001) referred to the case reported by Hamel et al. (1996) as one of cerebrooculofacioskeletal syndrome. The patient showed prenatal-onset growth deficiency, severe microcephaly, microphthalmia with no cataracts, cleft palate, cutaneous photosensitivity, and brain atrophy with no calcifications.
A number sign (#) is used with this entry because of evidence that cerebrooculofacioskeletal syndrome-4 (COFS4) is caused by homozygous or compound heterozygous mutation in the ERCC1 gene (126380) on chromosome 19q13. Description Cerebrooculofacioskeletal syndrome-4 is a severe autosomal recessive disorder characterized by growth retardation, dysmorphic facial features, arthrogryposis, and neurologic abnormalities. ... Echography did not reveal any congenital heart defects. Although other dysmorphology syndromes, such as Warburg Micro syndrome (600118) and Cockayne syndrome (see 216400), were considered, COFS was the preferred diagnosis because of the absence of genital abnormalities and retinopathy. ... This patient was diagnosed as having Cockayne syndrome on the basis of impaired RNA synthesis after UV radiation, indicating a defect in TC-NER. ... Molecular Genetics In a patient with cerebrooculofacioskeletal syndrome, Jaspers et al. (2007) detected compound heterozygosity for mutations in the ERCC1 gene.
Cockayne syndrome is a rare disease which causes short stature, premature aging ( progeria ), severe photosensitivity , and moderate to severe learning delay. This syndrome also includes failure to thrive in the newborn, very small head (microcephaly), and impaired nervous system development. ... There are three subtypes according to the severity of the disease and the onset of the symptoms: Cockayne syndrome type 1 (type A) , sometimes called “classic” or "moderate" Cockayne syndrome, diagnosed during early childhood Cockayne syndrome type 2 (type B) , sometimes referred to as the “severe” or "early-onset" type, presenting with growth and developmental abnormalities at birth Cockayne syndrome type 3 (type C) , a milder form of the disorder Cockayne syndrome is caused by mutations in either the ERCC8 (CSA) or ERCC6 (CSB) genes.
Cockayne syndrome type B (CSB; 133540) is an allelic disorder. ... Both were considered to have typical COFS syndrome. The girl died at age 5 years. ... Molecular Genetics Meira et al. (2000) presented evidence that 2 probands related to the Manitoba aboriginal population group within which COFS syndrome was originally reported by Lowry et al. (1971) and Pena and Shokeir (1974) had cellular phenotypes indistinguishable from those in Cockayne syndrome cells. ... In 3 unrelated patients with COFS syndrome, Laugel et al. (2008) identified biallelic mutations in the ERCC6 gene (see, e.g., 609413.0012-609413.0014). ... Nomenclature Shokeir (1982) suggested that there are two types of Pena-Shokeir syndrome: type I (208150), which shows multiple ankyloses, camptodactyly, facial anomalies and pulmonary hypoplasia (Pena and Shokeir (1974, 1976)); and type II, also known as the COFS syndrome.
A number sign (#) is used with this entry because of evidence that cerebrooculofacioskeletal syndrome-2 (COFS2) is caused by compound heterozygous mutation in the DNA repair gene XPD (ERCC2; 126340) on chromosome 19q13. ... For a phenotypic description and a discussion of genetic heterogeneity of cerebrooculofacioskeletal syndrome, see 214150. Clinical Features Graham et al. (2001) described a patient with COFS syndrome who was the child of nonconsanguineous Ashkenazi Jewish parents. ... Graham et al. (2001) proposed that patients with ultraviolet-sensitive COFS syndrome be included within the spectrum of individuals with impaired nucleotide-excision repair (NER) disorders.
Cockayne syndrome (CS) is a multisystem condition characterized by short stature, a characteristic facial appearance, premature aging, photosensitivity, progressive neurological dysfunction, and intellectual deficit. ... Subcutaneous lipoatrophy is present and can lead to signs of premature aging of the skin. COFS syndrome (see this term) is the extreme prenatal form of the CS clinical spectrum characterized by congenital microphthalmia and arthrogryposis.
Homozygous mutation in the ERCC5 gene can also cause cerebrooculofacioskeletal syndrome-3 (COFS3; 616570). Description For a general description of xeroderma pigmentosum, see XPA (278700), and of Cockayne syndrome, see CSA (216400). ... These patients were reported before the relationship between xeroderma pigmentosum group G and Cockayne syndrome was appreciated (Lalle et al., 2002). ... The mutant cells, unlike those from xeroderma pigmentosum group G/Cockayne syndrome patients, were capable of limited transcription-coupled repair of oxidative lesions. ... Genotype/Phenotype Correlations Some patients with a combined phenotype of xeroderma pigmentosum and Cockayne syndrome fall into complementation group G. ... Soltys et al. (2013) suggested that more severe ERCC5 defects that also impair the response to oxidative stress-induced injury, usually truncating mutations (see, e.g., 133530.0003), are associated with the more severe phenotype observed in Cockayne syndrome. History Complementation tests by cell fusion demonstrated that the NER syndromes are genetically heterogeneous and comprise 10 or more complementation groups: 7 in xeroderma pigmentosum, 2 in Cockayne syndrome, and 2 in TTD (Hoeijmakers, 1994).
A form of Antley-Bixler syndrome that includes disordered steroidogenesis (ABS1; 201750) is caused by mutation in the gene encoding cytochrome P450 oxidoreductase (POR; 124015). Description The Antley-Bixler syndrome (ABS) is an exceptionally rare craniosynostosis syndrome characterized by radiohumeral synostosis present from the perinatal period. ... Although the differential diagnosis included campomelic syndrome (see 114290), osteogenesis imperfecta (see 166200), and certain of the acrocephalosyndactyly syndromes (see 101200), the disorder appeared to be unique. ... Suzuki et al. (1987) described this syndrome in a brother and sister with first-cousin parents. ... Gorlin (1999) and Gripp et al. (1999) refuted the clinical diagnosis of Antley-Bixler syndrome and suggested that the patient had a nonspecific craniosynostosis syndrome.
Antley Bixler syndrome is a rare condition that is primarily characterized by craniofacial abnormalities and other skeletal problems. ... The exact underlying cause of Antley Bixler syndrome is unknown in many cases; however, some are due to changes (mutations) in the FGFR2 gene or the POR gene.
A very rare disorder characterised by craniosynostosis with midface hypoplasia, radiohumeral synostosis, femoral bowing and joint contractures. Epidemiology It has been described in more than 30 patients. Clinical description Children present with characteristic facial features, including a large domed forehead, flat nose, and midface hypoplasia with proptosis and dysplastic ears. Arachnodactyly and/or camptodactyly have also been reported. A diverse range of malformations (cardiac, anal or vertebral) are often associated. Urogenital anomalies with sexual ambiguity due to impaired steroidogenesis can occur. Intellectual development is variable. Differential diagnosis A similar clinical picture is observed in patients exposed in utero to fluconazole, a lanosterol 14 alpha-demethylase inhibitor.
A number sign (#) is used with this entry because Griscelli syndrome type 3 (GS3), which is characterized by hypomelanosis with no immunologic or neurologic manifestations, can be caused by mutation in the melanophilin (MLPH; 606526) or MYO5A (160777) genes. For a discussion of phenotypic and genetic heterogeneity in Griscelli syndrome, see the entry for GS1 (214450). Clinical Features Sanal et al. (2002) reported 2 patients with silver-gray hair, eyebrows, and eyelashes who had large clumps of pigment, typical of Griscelli syndrome, on microscopic analysis of their hair shafts. ... INHERITANCE - Autosomal recessive HEAD & NECK Eyes - Silver-gray eyelashes - Silver-gray eyebrows SKIN, NAILS, & HAIR Hair - Silver-gray hair - Silver-gray eyelashes - Silver-gray eyebrows - Large clumps of pigment irregularly distributed along hair shaft (light microscopy) NEUROLOGIC - No neurologic abnormalities IMMUNOLOGY - No immunologic abnormalities MISCELLANEOUS - Genetic heterogeneity - See also Griscelli syndrome, type 1 ( 214450 ) for a similar disorder with characteristic neurologic disease and Griscelli syndrome, type 2 ( 607624 ) for a similar disorder with characteristic immunodeficiency/hemophagocytic syndrome.
Trisomy 22 is a chromosome disorder in which an extra (third) copy of chromosome 22 is present in every cell of the body where there should normally only be two copies. This condition is commonly found in miscarriages, but only rarely in liveborn infants. Most affected individuals die shortly before or shortly after birth due to severe complications. Common features include an underdeveloped midface (midface hypoplasia) with flat/broad nasal bridge, malformed ears with pits or tags, cleft palate, hypertelorism (wide-spaced eyes), microcephaly and other cranial abnormalities, congenital heart disease , genital abnormalities, and intrauterine growth restriction (IUGR).
Boy age seven years with Pitt-Hopkins syndrome (same individual as in Figure 1). ... Boy age 13 years with Pitt-Hopkins syndrome (same individual as in Figures 1 and 2). ... Woman age 24 years with Pitt-Hopkins syndrome (same individual as in Figure 4). ... See Joubert Syndrome to view genes associated with this phenotype. 2. Joubert syndrome is inherited predominantly in an autosomal recessive manner.
A number sign (#) is used with this entry because of evidence that Pitt-Hopkins syndrome (PTHS) is caused by heterozygous mutation in the TCF4 gene (602272) on chromosome 18q21. ... See also Pitt-Hopkins-like syndrome-1 (610042), caused by mutation in the CNTNAP2 gene (604569) on chromosome 7q35, and Pitt-Hopkins-like syndrome-2 (600565), caused by mutation in the NRXN1 gene (600565) on chromosome 2p16.3. ... They further defined the Pitt-Hopkins syndrome phenotype with a description of 2 new patients. ... Among their newly identified patients, all had features consistent with Pitt-Hopkins syndrome, although only 3 had breathing anomalies and none had seizures. ... Marangi et al. (2011) identified haploinsufficiency for the TCF4 gene in 14 of 63 Italian patients referred for suspicion of Pitt-Hopkins syndrome. One patient with the full syndrome had a balanced translocation involving the TCF4 gene.
Pitt-Hopkins syndrome (PTHS) is a genetic syndrome that causes developmental delays, moderate to severe intellectual disability, behavioral differences, distinctive facial features, and breathing problems such as episodes of rapid breathing ( hyperventilation ) and breath-holding.
A rare multiple congenital anomalies syndrome characterized by the association of intellectual deficit, characteristic facial morphology and problems of abnormal and irregular breathing. ... Underdevelopment of external and internal reproductive organs (small penis, cryptorchidism, labial fusions) occurs regularly in males and females. Etiology The syndrome is caused by heterozygous, usually de novo mutations in the TCF4 gene (18q21), coding for a ubiquitous b-HLH transcription factor. ... Differential diagnosis The principal differential diagnosis includes Angelman syndrome, Rett syndrome and Mowat-Wilson syndrome. ... Developmental assessments are needed to tailor medical services to each individual's needs. The use of syndrome‐specific information booklets is recommended for affected families and caregivers; adequate information regarding the breathing disturbances is especially important to avoid mismanagement.
Pitt-Hopkins syndrome is a condition characterized by intellectual disability and developmental delay, breathing problems, recurrent seizures (epilepsy), and distinctive facial features. People with Pitt-Hopkins syndrome have moderate to severe intellectual disability. ... Some older individuals with Pitt-Hopkins syndrome develop widened and rounded tips of the fingers and toes (clubbing) because of recurrent episodes of decreased oxygen in the blood. ... Epilepsy occurs in most people with Pitt-Hopkins syndrome and usually begins during childhood, although it can be present from birth. ... Frequency Pitt-Hopkins syndrome is thought to be a very rare condition.