-
Congenital Myasthenic Syndromes
Mayo_clinic
Overview Congenital myasthenic syndromes are rare hereditary (genetic) conditions resulting from a defect at the junction where your nerve stimulates muscle activity. That defect causes muscle weakness. Congenital myasthenic syndromes may affect your nerve cells (presynaptic), your muscle cells (postsynaptic), or the space between your nerve and muscle cells (synaptic).GFPT1, AGRN, SCN4A, SLC18A3, SLC5A7, ALG2, LRP4, CHRND, ALG14, COLQ, RAPSN, DOK7, GMPPB, VAMP1, DPAGT1, MUSK, COL13A1, PLEC, LAMB2, PREPL, SNAP25, SYT2, CHRNG, CHRNB1, CHRNE, CHAT, TAPBPL, ACHE, C17orf107, CHRNA1, CD2AP, BCHE, HNRNPH1, HNRNPH2, MYO9A, DAP, EIF3K, SRSF1, UTRN, NGFR, NTRK1, CAV1, SMN1, SMN2, DES, ERBB3, SEA, CHD7, LAMA5, SEMA7A, TPM3, B3GAT1, SLC25A1, RPH3A, VCP, DNAJA3, MACF1
-
Favre–racouchot Syndrome
Wikipedia
Favre–Racouchot syndrome Other names Favre–Racouchot disease , [1] and Nodular cutaneous elastosis with cysts and comedones [1] Affected eyelids and upper cheeks with greyish discolouration. Specialty Dermatology Favre–Racouchot syndrome is a solar elastotic disorder consisting of multiple open comedones that occurs in skin damaged by sunlight, especially under and lateral of the eyes. ... Topical retinoids , Isotretinoin , curettage , and dermabrasion have also been used with some success. [3] Eponym [ edit ] Favre–Racouchot syndrome is named after the French dermatologist Maurice Favre and his pupil Jean Racouchot (1908-1994). ... New England Journal of Medicine . 366 (16). pp. e25. doi : 10.1056/NEJMicm1104059 . ^ "Favre-Racouchot Syndrome - American Osteopathic College of Dermatology (AOCD)" . www.aocd.org . Retrieved 2019-09-01 . ^ "Favre-Racouchot syndrome" . External links [ edit ] Classification D ICD - 10 : L57.8 ( ILDS L57.820) ICD - 9-CM : 701.8 External resources eMedicine : article/1119362 v t e Radiation-related disorders / Photodermatoses Ultraviolet / ionizing Sunburn Phytophotodermatitis Solar urticaria Polymorphous light eruption Benign summer light eruption Juvenile spring eruption Acne aestivalis Hydroa vacciniforme Solar erythema Non-ionizing Actinic rays Actinic keratosis Atrophic actinic keratosis Hyperkeratotic actinic keratosis Lichenoid actinic keratosis Pigmented actinic keratosis Actinic cheilitis Actinic granuloma Actinic prurigo Chronic actinic dermatitis Infrared / heat Erythema ab igne ( Kangri ulcer Kairo cancer Kang cancer Peat fire cancer ) Cutis rhomboidalis nuchae Poikiloderma of Civatte Other Radiation dermatitis Acute Chronic radiodermatitis ) Favre–Racouchot syndrome Photoaging Photosensitivity with HIV infection Phototoxic tar dermatitis This cutaneous condition article is a stub .

-
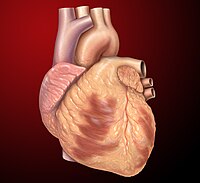
Carditis
Wikipedia
CS1 maint: others ( link ) v t e Cardiovascular disease (heart) Ischaemic Coronary disease Coronary artery disease (CAD) Coronary artery aneurysm Spontaneous coronary artery dissection (SCAD) Coronary thrombosis Coronary vasospasm Myocardial bridge Active ischemia Angina pectoris Prinzmetal's angina Stable angina Acute coronary syndrome Myocardial infarction Unstable angina Sequelae hours Hibernating myocardium Myocardial stunning days Myocardial rupture weeks Aneurysm of heart / Ventricular aneurysm Dressler syndrome Layers Pericardium Pericarditis Acute Chronic / Constrictive Pericardial effusion Cardiac tamponade Hemopericardium Myocardium Myocarditis Chagas disease Cardiomyopathy Dilated Alcoholic Hypertrophic Tachycardia-induced Restrictive Loeffler endocarditis Cardiac amyloidosis Endocardial fibroelastosis Arrhythmogenic right ventricular dysplasia Endocardium / valves Endocarditis infective endocarditis Subacute bacterial endocarditis non-infective endocarditis Libman–Sacks endocarditis Nonbacterial thrombotic endocarditis Valves mitral regurgitation prolapse stenosis aortic stenosis insufficiency tricuspid stenosis insufficiency pulmonary stenosis insufficiency Conduction / arrhythmia Bradycardia Sinus bradycardia Sick sinus syndrome Heart block : Sinoatrial AV 1° 2° 3° Intraventricular Bundle branch block Right Left Left anterior fascicle Left posterior fascicle Bifascicular Trifascicular Adams–Stokes syndrome Tachycardia ( paroxysmal and sinus ) Supraventricular Atrial Multifocal Junctional AV nodal reentrant Junctional ectopic Ventricular Accelerated idioventricular rhythm Catecholaminergic polymorphic Torsades de pointes Premature contraction Atrial Junctional Ventricular Pre-excitation syndrome Lown–Ganong–Levine Wolff–Parkinson–White Flutter / fibrillation Atrial flutter Ventricular flutter Atrial fibrillation Familial Ventricular fibrillation Pacemaker Ectopic pacemaker / Ectopic beat Multifocal atrial tachycardia Pacemaker syndrome Parasystole Wandering atrial pacemaker Long QT syndrome Andersen–Tawil Jervell and Lange-Nielsen Romano–Ward Cardiac arrest Sudden cardiac death Asystole Pulseless electrical activity Sinoatrial arrest Other / ungrouped hexaxial reference system Right axis deviation Left axis deviation QT Short QT syndrome T T wave alternans ST Osborn wave ST elevation ST depression Strain pattern Cardiomegaly Ventricular hypertrophy Left Right / Cor pulmonale Atrial enlargement Left Right Athletic heart syndrome Other Cardiac fibrosis Heart failure Diastolic heart failure Cardiac asthma Rheumatic fever This article about a medical condition affecting the circulatory system is a stub .
-
Joubert Syndrome 24
Omim
A number sign (#) is used with this entry because of evidence that Joubert syndrome-24 (JBTS24) is caused by homozygous mutation in the TCTN2 gene (613846) on chromosome 12q24. Description Joubert syndrome-24 is an autosomal recessive ciliopathy characterized by delayed psychomotor development associated with cerebellar hypoplasia manifest as the molar tooth sign on brain imaging. ... For a phenotypic description and a discussion of genetic heterogeneity of Joubert syndrome, see 213300. Clinical Features Sang et al. (2011) reported 7 patients from 4 unrelated families with Joubert syndrome. ... Huppke et al. (2015) reported a 7.5-year-old Turkish boy, born of consanguineous parents, with a neurodevelopmental disorder consistent with Joubert syndrome. At birth, the patient was noted to have postaxial hexadactyly of all 4 extremities, but no other dysmorphic features. ... In a comprehensive study of 279 patients from 232 unrelated families with Joubert syndrome in whom a genetic basis was determined by molecular analysis of 27 candidate genes, Bachmann-Gagescu et al. (2015) found a significant association between mutations in the TCTN2 gene and encephalocele (odds ratio (OR) of 13.6) and polydactyly (OR of 18.7).
-
Multiple Synostoses Syndrome 4
Omim
A number sign (#) is used with this entry because of evidence that multiple synostoses syndrome-4 (SYNS4) is caused by heterozygous mutation in the GDF6 gene (601147) on chromosome 8q22. Description Multiple synostoses syndrome-4 is characterized by fusion of carpal and tarsal bones, as well as conductive hearing loss (Terhal et al., 2018). For a general phenotypic description and a discussion of genetic heterogeneity of multiple synostoses syndrome, see SYNS1 (186500). Clinical Features Wang et al. (2016) reported a large 6-generation Chinese family in which 46 members experienced joint fusions of variable severity. ... Terhal et al. (2018) studied 5 of the 10 affected members of a 4-generation family segregating autosomal dominant multiple synostoses syndrome resulting in tarsal and carpal bone fusions and variable hearing loss. ... In a 4-generation family with multiple synostoses syndrome, negative for mutation in 2 known SYNS-associated genes, Terhal et al. (2018) sequenced the candidate gene GDF6 and identified heterozygosity for a missense mutation (S429R; 601147.0011) that segregated with disease and was not found in public variant databases.
-
Usher Syndrome, Type Iv
Omim
A number sign (#) is used with this entry because of evidence that Usher syndrome type IV (USH4) is caused by homozygous mutation in the ARSG gene (610008) on chromosome 17q24. Description An atypical form of Usher syndrome, here designated type IV, is an autosomal recessive disorder characterized by late onset of retinitis pigmentosa and usually late-onset of progressive sensorineural hearing loss without vestibular involvement (summary by Khateb et al., 2018). For a discussion of genetic heterogeneity of Usher syndrome, see 276900. Clinical Features Khateb et al. (2018) described 5 patients from 3 Yemenite Jewish families (MOL0120, MOL0737, and TB55) with an atypical form of Usher syndrome. ... Mapping By homozygosity mapping followed by whole-genome and whole-exome sequencing in 3 Yemenite Jewish families segregating an atypical form of Usher syndrome, Khateb et al. (2018) identified a single shared homozygous 66- to 69.4-Mb region on chromosome 17. Molecular Genetics In 5 affected members of 3 consanguineous Yemenite Jewish families with an atypical form of Usher syndrome (618144), Khateb et al. (2018) identified homozygosity for a missense mutation in the ARSG gene (D45Y; 610008.0001).
-
Multicentric Carpotarsal Osteolysis Syndrome
Wikipedia
Multicentric carpotarsal osteolysis syndrome Other names MCTO [1] This condition is inherited in an autosomal dominant manner. Specialty Medical genetics Multicentric carpotarsal osteolysis syndrome (MCTO) is a rare autosomal dominant condition. [2] This condition is also known as idiopathic multicentric osteolysis with nephropathy. ... Classification [ edit ] This condition has been classified into five types. [4] Type 1: hereditary multicentric osteolysis with dominant transmission Type 2: hereditary multicentric osteolysis with recessive transmission Type 3: nonhereditary multicentric osteolysis with nephropathy Type 4: Gorham–Stout syndrome Type 5: Winchester syndrome – defined as a monocentric disease with autosomal recessive inheritance Treatment [ edit ] Optimal treatment for this condition is unclear. ... History [ edit ] This condition was first described by Shurtleff et al. in 1964. [2] References [ edit ] ^ "OMIM Entry - # 166300 - MULTICENTRIC CARPOTARSAL OSTEOLYSIS SYNDROME; MCTO" . omim.org . Retrieved 27 April 2019 . ^ a b Shurtleff DB, Sparkes RS, Clawson DK, Guntheroth WG, Mottet NK (1964) Hereditary osteolysis with hypertension and nephropathy. ... Am J Hum Genet 90: 494-501 ^ Hardegger F, Simpson LA, Segmueller G (1985) The syndrome of idiopathic osteolysis. Classification, review, and case report.

-
Limber Tail Syndrome
Wikipedia
Please help to improve this article by introducing more precise citations. ( August 2009 ) ( Learn how and when to remove this template message ) A Labrador Retriever with Limber Tail Syndrome. Limber tail syndrome , or acute caudal myopathy , is a disorder of the muscles in the tail, usually affecting working dogs . [1] It is an injury occurring mostly in sporting or working dogs such as English Pointers , English Setters , Foxhounds , Beagles , and Labrador Retrievers . Limber tail syndrome is also known as swimmer's tail, cold water tail, broken tail, dead tail, "happy tail" or broken wag. ... Treatment [ edit ] With rest, the tail returns to normal within a few days. [2] Pain relief, such as a nonsteroidal anti-inflammatory drug may be administered. [2] The symptoms may recur. [2] References [ edit ] ^ De Lahunta, Alexander; Glass, Eric (2009). "Limber tail syndrome, or acute caudal myopathy". Veterinary Neuroanatomy and Clinical Neurology (3rd ed.). ... Steiss, Janet E. & Wright, J.C., Limber Tail Syndrome in Hunting Dogs, Sports Medicine Program Newsletter, Auburn University College of Veterinary Medicine, Winter 1995 Grayson, Peggy, Water and the dead tail syndrome, Dog World, May 5, 1995 Steiss, J; Braund, K; Wright, J; Lenz, S; Hudson, J; Brawner, W; Hathcock, J; Purohit, R; Bell, L; Horne, R (1999).

-
Medich Giant Platelet Syndrome
Orphanet
Medich giant platelet syndrome (MGPS) is a platelet granule disorder characterized by thrombocytopenia with giant platelets resulting in easy bleeding.
-
3p25.3 Microdeletion Syndrome
Orphanet
3p25.3 microdeletion syndrome is a rare chromosomal anomaly characterized by intellectual disability, epilepsy or EEG abnormalities, poor speech, ataxia, and stereotypic hand movements.
-
Congenital Amputation
Wikipedia
It is known to be caused by blood clots forming in the fetus while in utero (vascular insult) and from amniotic band syndrome : fibrous bands of the amnion that constrict foetal limbs to such an extent that they fail to form or actually fall off due to missing blood supply. ... One common cause is amniotic band syndrome , which occurs when the inner fetal membrane ( amnion ) ruptures without injury to the outer membrane ( chorion ). ... Walter JH, Goss LR, Lazzara AT (July–August 1998). "Amniotic band syndrome". The Journal of Foot and Ankle Surgery . 37 (4): 325–33. doi : 10.1016/s1067-2516(98)80070-7 . ... Light TR, Ogden JA (May–June 1993). "Congenital constriction band syndrome. Pathophysiology and treatment" . ... The Free Dictionary . v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum
-
Visceroptosis
Wikipedia
Frantz Glénard (1848–1920) Visceroptosis is a known risk factor for the development of Superior mesenteric artery syndrome . Visceroptosis also is known as Glénard's disease (after French physician Frantz Glénard [1848–1920]). ... Worcester Categories Corsetry Fashion Foundation garments v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum
-
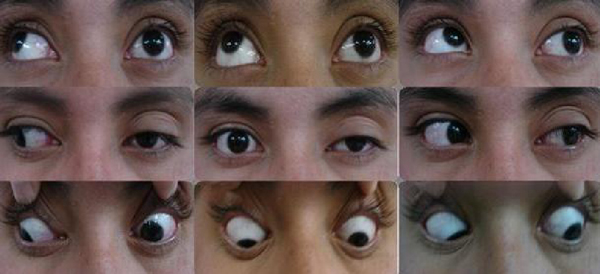
Ophthalmoparesis
Wikipedia
The muscle , as in progressive external ophthalmoplegia or Kearns–Sayre syndrome . The neuromuscular junction , as in myasthenia gravis . The relevant cranial nerves (specifically the oculomotor , trochlear , and abducens ), as in cavernous sinus syndrome or raised intracranial pressure . The brainstem nuclei of these nerves, as in certain patterns of brainstem stroke such as Foville's syndrome . White matter tracts connecting these nuclei, as in internuclear ophthalmoplegia , an occasional finding in multiple sclerosis . Dorsal midbrain structures, as in Parinaud's syndrome . Certain parts of the cerebral cortex (including the frontal eye fields ), as in stroke . ... Thiamine deficiency can cause ophthalmoparesis in susceptible persons; this is part of the syndrome called Wernicke encephalopathy .MGME1, NPC1, POLG, CHRNE, TWNK, TRNF, RAPSN, TRNH, TRNK, TRNL1, TRNQ, TRNS1, TRNS2, TRNV, TRNW, MUSK, SCN4A, ATXN1, ND6, SLC25A1, COLQ, TCIRG1, AFG3L2, PTPN22, SLC5A7, AK9, DOK7, AGRN, CACNA1S, KCNJ18, ND5, LAMB2, CHAT, CHRNA1, CHRNB1, CHRND, COL13A1, DES, ATN1, GABRA3, GFPT1, KRT5, ND4, KRT14, COX1, COX3, ND3, ND2, MAPT, ND1, ATP6, LRP4, COX2, MTM1, ATXN2, CHMP1B, TYMP, SCO2, TGFB1, LY6E, CACNA1A
-
Microstomia
Wikipedia
Contents 1 Congenital 2 Acquired 3 References 4 External links Congenital [ edit ] It is a feature of many craniofacial syndromes , including Freeman–Sheldon syndrome and Sheldon-Hall syndromes (or distal arthrogryposis multiplex congenita ). It may present with whistling-face feature, as well, as in Freeman-Sheldon syndrome. In this syndrome, it impairs alimentation and may require repeated oral surgeries (called commissurotomy ) to improve function. [ citation needed ] Acquired [ edit ] Microstomia can occur as a result of scarring due to many conditions. ... External links [ edit ] Classification D ICD - 10 : Q18.5 ICD - 9-CM : 744.84 MeSH : D008865 DiseasesDB : 29609 SNOMED CT : 14582003 External resources eMedicine : ent/148 v t e Congenital malformations and deformations of face and neck Face jaw : Otocephaly mouth : Macrostomia Microstomia lip : Macrocheilia Microcheilia chin : Microgenia multiple/other: Hallermann–Streiff syndrome Branchial cleft cyst Neck Webbed neck Ungrouped Preauricular sinus and cystABL1, MAPRE2, AKT3, HUWE1, PQBP1, ZMPSTE24, SPEG, SEPTIN9, SRCAP, TXNL4A, TLK2, PIGN, POLR3A, B4GALT7, CHSY1, POGZ, MAPK8IP3, CAMTA1, SATB2, CLCF1, MED12, AMMECR1, EIF4A3, SEMA3E, ZBTB16, BRPF1, CDC45, KCNAB2, CDK13, TP63, DCHS1, CRLF1, TRIP4, COG1, RECQL4, CHST3, TBX4, ADAMTS3, POLR1C, SEC24C, TTC37, ATP6V0A2, ORC6, AKT1, SP7, PRDM16, BCL11B, FAT4, FBXO11, CDT1, UBE3B, COG7, CHST14, CANT1, RTTN, TBC1D20, GSC, CCBE1, TUBB, JMJD1C, PHACTR1, NALCN, RSPO2, PIEZO2, TRMT5, GATAD2B, FAM20C, SIN3A, SH2B1, AUTS2, ZBTB20, B3GAT3, DCPS, DSE, SLC25A24, DONSON, GMNN, POLR1D, RLIM, RIPK4, IMPAD1, CHD7, OSGEP, IARS2, PCGF2, YWHAE, WNT3, GP1BB, BPTF, FBN1, FGFR2, FLNA, GABRD, GBA, GJA1, GNAI3, HIVEP2, UFD1, HSPG2, IGF1R, ITGA3, LAMA3, LAMB3, LAMC2, LMNA, LTBP3, EP300, EDN1, DHCR24, CRKL, BIN1, APC, ARVCF, RERE, BCR, BMP2, BMPR1A, CCND2, CDC6, CHRNG, COL2A1, COL3A1, COL7A1, COL11A1, COL11A2, COMT, CREBBP, SMAD4, MAF, MCM5, TAF6, PTEN, ALDH18A1, RPL10, RREB1, RYR1, SKI, SMS, SON, TBX1, MAPK1, TCOF1, TNNI2, TNNT3, TPM2, TTN, TTPA, HIRA, TWIST1, PTCH1, POLD1, MECP2, ORC1, MMP1, MUSK, MYBPC1, MYH3, NFIA, NFIX, NONO, NOTCH2, ORC4, PRRX1, OTX2, PAFAH1B1, PAX3, PIGA, PIK3CA, PIK3R2, PLCB4, FREM2, BARX1
-
Eosinophilic Dermatosis
Wikipedia
Retrieved 1 May 2011 . v t e Neutrophilic and eosinophilic dermatoses Eosinophilic dermatosis With vasculitis Eosinophilic vasculitis Eosinophilic granulomatosis with polyangiitis Without vasculitis Arthropod assault Eosinophilic cellulitis Hypereosinophilic syndrome Papuloerythroderma of Ofuji Granuloma faciale Eosinophilic folliculitis Ungrouped Angiolymphoid hyperplasia with eosinophilia / Kimura's disease Annular erythema of infancy Eosinophilic fasciitis Eosinophilic granuloma Eosinophilic ulcer of the oral mucosa Erythema toxicum neonatorum Incontinentia pigmenti Itchy red bump disease Juvenile xanthogranuloma Pachydermatous eosinophilic dermatitis Papular eruption of blacks Pruritic papular eruption of HIV disease Reactive neutrophilic dermatoses Epidermis Keratoderma blennorrhagicum Subcorneal pustular dermatosis Dermis without vasculitis : Sweet's syndrome Pyoderma gangrenosum Bowel-associated dermatosis–arthritis syndrome with vasculitis: Neutrophilic dermatosis of the dorsal hands Ungrouped Acute erythema nodosum Marshall syndrome Neutrophilic eccrine hidradenitis Pyogenic arthritis–pyoderma gangrenosum–acne syndrome Rheumatoid neutrophilic dermatitis Superficial granulomatous pyoderma Sweet's syndrome-like dermatosis Vesicopustular dermatosis
-
Hagemoser–weinstein–bresnick Syndrome
Wikipedia
Hagemoser–Weinstein–Bresnick syndrome Hagemoser–Weinstein–Bresnick syndrome is inherited in an autosomal dominant manner Specialty Ophthalmology , otorhinolaryngology Hagemoser–Weinstein–Bresnick syndrome is an autosomal dominant genetic disorder first described by Hagemoser et al. in 1989.
-
Pituitary Acth Hypersecretion
Wikipedia
Pituitary ACTH hypersecretion Specialty Endocrinology Pituitary ACTH hypersecretion (or Cushing disease ) is a form of hyperpituitarism characterized by an abnormally high level of ACTH produced by the anterior pituitary . [1] It is one of the causes of Cushing's syndrome . (However, Cushing's syndrome can be caused by many other causes, including exogenous administration.) ... External links [ edit ] Classification D ICD - 10 : E24.0 ICD - 9-CM : 255.0 MeSH : D047748 v t e Pituitary disease Hyperpituitarism Anterior Acromegaly Hyperprolactinaemia Pituitary ACTH hypersecretion Posterior SIADH General Nelson's syndrome Hypophysitis Hypopituitarism Anterior Kallmann syndrome Growth hormone deficiency Hypoprolactinemia ACTH deficiency / Secondary adrenal insufficiency GnRH insensitivity FSH insensitivity LH/hCG insensitivity Posterior Neurogenic diabetes insipidus General Empty sella syndrome Pituitary apoplexy Sheehan's syndrome Lymphocytic hypophysitis Pituitary adenoma This article about an endocrine, nutritional, or metabolic disease is a stub .USP8, POMC, PPARG, AIP, CDH23, SCG5, SST, NR3C1, GNAI2, CRH, EGFR, GH1, MEN1, GHRL, H3P10, CDKN2A, IL6, GNAS, HSD11B2, HSP90AA1, CABLES1, TBX19, MC2R, AVPR1B, NR2C2, SSTR5, SSTR2, PCSK1, SSTR1, PRKAR1A, SMARCA4, PRL, BGLAP, HDAC2, USP48, CYP11B1, GBA3, GIP, GHSR, CDKN1B, DICER1, BRAF, SMUG1, E2F1, RNU1-1, SERPINA6, SP1, MOCOS, PRSS55, MIR449C, SPZ1, LINC02210-CRHR1, SMS, SLC20A1, SLC8A1, ZC4H2, TP73, STAT3, THBS1, TP53, SFRP2, CRISP2, CD274, VDR, VIM, RSS, LPXN, MED7, CIB1, CHEK2, ARHGAP24, ABL1, RNU1-4, COX8A, NR4A1, GHR, FGFR4, EPHB2, EGF, DRD2, CRHR1, CRHBP, CNC2, HSF1, CIRBP, CD14, BMP4, BCL2, CCND1, KLK3, APOB, APOA1, HSD11B1, IGF1, BRD2, CNTN3, MAP2K7, MAPK1, PRKACA, PPARA, POU1F1, PCSK2, AGRP, PAX7, SERPINB2, IL1B, SERPINE1, NEUROD1, NDUFA2, MSH2, MGMT, SMCP, LIFR, KRT5, PCNA
-
Woods Syndrome
Omim
Woods et al. (1992) concluded that the condition in their family was different from Filippi syndrome (272440) because the facial appearances were different and children with Filippi syndrome do not have localizing neurologic signs. For a similar craniofacial syndrome, see 251255. Inheritance Consanguinity in the family reported by Woods et al. (1992) suggested autosomal recessive inheritance of the disorder.
-
Adrenalitis
Wikipedia
External links [ edit ] Classification D ICD - 10 : E27.8 v t e Adrenal gland disorder Hyperfunction Aldosterone Hyperaldosteronism Primary aldosteronism Conn syndrome Bartter syndrome Glucocorticoid remediable aldosteronism AME Liddle's syndrome 17α CAH Pseudohypoaldosteronism Cortisol Cushing's syndrome Pseudo-Cushing's syndrome Steroid-induced osteoporosis Sex hormones 21α CAH 11β CAH Hypofunction Aldosterone Hypoaldosteronism 21α CAH 11β CAH Cortisol CAH Lipoid 3β 11β 17α 21α Sex hormones 17α CAH Inborn errors of steroid metabolism Adrenal insufficiency Adrenal crisis Adrenalitis Xanthogranulomatous Addison's disease Waterhouse–Friderichsen syndrome This article about an endocrine, nutritional, or metabolic disease is a stub .
-
Stiff Person Syndrome
Gard
Stiff person syndrome (SPS) is a rare, progressive syndrome that affects the nervous system , specifically the brain and spinal cord. ... Persistent symptoms can lead to abnormal posturing of the spine, such as being hunched over. The syndrome affects twice as many women as men.





