Unsourced material may be challenged and removed. Find sources: "Sengers syndrome" – news · newspapers · books · scholar · JSTOR ( April 2018 ) ( Learn how and when to remove this template message ) Senger's syndrome Autosomal recessive pattern is the inheritance manner of this condition Causes Mutations in the AGK and SLC25A4 genes Sengers syndrome is a rare autosomal recessive condition characterised by congenital cataract , hypertrophic cardiomyopathy , muscle weakness and lactic acidosis after exercise. ... Differential diagnosis [ edit ] Isolated ATP synthase deficiency Barth syndrome TMEM70 deficiency Treatment [ edit ] Surgery for the cataracts may be needed. ... Because of its rarity the prognosis for the chronic form is not well established but survival into adulthood has been reported. Epidemiology [ edit ] Sengers syndrome is a rare disorder. About 40 cases have been reported worldwide. [ citation needed ] History [ edit ] This condition was first described in 1975. [1] References [ edit ] ^ Sengers RCA, ter Haar, BGA, Trijbels JMF, Willems JL, Daniels O, Stadhouders AM (1975) Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise.
A number sign (#) is used with this entry because Sengers syndrome, also known as cardiomyopathic mitochondrial DNA depletion syndrome-10 (MTDPS10), is caused by homozygous or compound heterozygous mutation in the AGK gene (610345) on chromosome 7q34. For a discussion of genetic heterogeneity of autosomal recessive mtDNA depletion syndromes, see MTDPS1 (603041). Description Sengers syndrome is an autosomal recessive mitochondrial disorder characterized by congenital cataracts, hypertrophic cardiomyopathy, skeletal myopathy, exercise intolerance, and lactic acidosis. ... The authors noted that the phenotype was more severe than usually reported in Sengers syndrome, and suggested a mitochondrial defect. ... Mayr et al. (2012) reported 5 patients, including 2 sibs, with Sengers syndrome and provided follow-up of 7 previously reported patients. ... Thus, they proposed that 'transcriptional, translational, or posttranslational events' are responsible for the ANT1 deficiency associated with Sengers syndrome. Molecular Genetics In 2 unrelated patients with myopathic mtDNA depletion syndrome, Calvo et al. (2012) identified homozygous or compound heterozygous mutations in the AGK gene (610345.0001-610345.0003).
Differential diagnosis Differential diagnoses include mitochondrial encephalo-cardio-myopathy due to TMEM70 deficiency, isolated ATP synthase deficiency and Barth syndrome (see these terms). Antenatal diagnosis Prenatal genetic testing may be possible for families with affected children.
A number sign (#) is used with this entry because of evidence that autosomal recessive mitochondrial DNA depletion syndrome-12B (MTDPS12B) is caused by homozygous or compound heterozygous mutation in the SLC25A4 gene (103220), which encodes ANT1, on chromosome 4q35. ... Description Mitochondrial DNA depletion syndrome-12B is an autosomal recessive mitochondrial disorder characterized by childhood onset of slowly progressive hypertrophic cardiomyopathy and generalized skeletal myopathy resulting in exercise intolerance, and, in some patients, muscle weakness and atrophy. ... For a discussion of genetic heterogeneity of mtDNA depletion syndromes, see MTDPS1 (603041). Clinical Features Bakker et al. (1993) described an 8-year-old boy with adenine nucleotide translocator deficiency in muscle who was first investigated at the age of 3.5 years because of shortness of breath and rapid fatigue.
A number sign (#) is used with this entry because of evidence that autosomal dominant Adams-Oliver syndrome-3 (AOS3) can be caused by heterozygous mutation in the RBPJ gene (147183) on chromosome 4p15. Description Hassed et al. (2012) described an autosomal dominant form of Adams-Oliver syndrome involving characteristic vertex scalp defects and terminal limb defects, but without congenital heart defects, other associated defects, or immune defects. For a discussion of genetic heterogeneity of Adams-Oliver syndrome, see AOS1 (100300). Clinical Features Hassed et al. (2012) reported 2 families segregating autosomal dominant Adams-Oliver syndrome. ... Molecular Genetics Using a variant-filtering strategy to perform exome resequencing in 2 unrelated families with Adams-Oliver syndrome, Hassed et al. (2012) identified 2 different heterozygous missense mutations in the RBPJ gene (147183.0001 and 147183.0002) that segregated with disease in each family.
A number sign (#) is used with this entry because of evidence that Adams-Oliver syndrome-6 (AOS6) is caused by heterozygous mutation in the DLL4 gene (605185) on chromosome 15q15. Description Adams-Oliver syndrome is a rare developmental disorder defined by the combination of aplasia cutis congenita of the scalp vertex and terminal transverse limb defects (e.g., amputations, syndactyly, brachydactyly, or oligodactyly). ... For a discussion of genetic heterogeneity of Adams-Oliver syndrome, see AOS1 (100300). Molecular Genetics Meester et al. (2015) screened 91 families with Adams-Oliver syndrome for mutations in the candidate gene DLL4 and identified heterozygous nonsense and missense mutations in 9 families (see, e.g., 605185.0001-605185.0006).
A number sign (#) is used with this entry because of evidence that Adams-Oliver syndrome-5 (AOS5) is caused by heterozygous mutation in the NOTCH1 gene (190198) on chromosome 9q34. Description Adams-Oliver syndrome (AOS) is a rare developmental disorder defined by the combination of aplasia cutis congenita of the scalp vertex and terminal transverse limb defects (e.g., amputations, syndactyly, brachydactyly, or oligodactyly). ... For a discussion of genetic heterogeneity of Adams-Oliver syndrome, see AOS1 (100300). Clinical Features Dallapiccola et al. (1992) observed aplasia cutis congenita and coarctation of the aorta in a mother and son. ... Vandersteen and Dixon (2011) described a father and 2 daughters from New Zealand with Adams-Oliver syndrome. The proband was a 24-year-old woman with congenital prominent scalp veins, cutis marmorata telangiectatica congenita (CMTC; 219250), and brachydactyly. ... Molecular Genetics Stittrich et al. (2014) performed whole-exome sequencing in affected individuals from 11 families with Adams-Oliver syndrome who were negative for mutations in known AOS-associated genes.
A number sign (#) is used with this entry because of evidence that autosomal recessive Adams-Oliver syndrome-4 (AOS4) can be caused by homozygous mutation in the EOGT gene (614789) on chromosome 3p14. Description Adams-Oliver syndrome is a rare congenital disorder characterized by aplasia cutis congenita and terminal transverse limb defects. ... For a discussion of genetic heterogeneity of Adams-Oliver syndrome (AOS), see AOS1 (100300). Clinical Features Shaheen et al. (2013) studied 5 affected children from 3 consanguineous Arab families who had Adams-Oliver syndrome. ... Mapping In affected members of 3 consanguineous Arab families with Adams-Oliver syndrome in whom no mutation in the DOCK6 gene (614194) was detected, Shaheen et al. (2013) found autozygome overlap on a previously unreported 2,744,933-bp locus at Chr3:66,612,406-69,357,338 (GRCh37).
Description Adams-Oliver syndrome-2 is an autosomal recessive multiple congenital anomaly syndrome characterized by aplasia cutis congenita (ACC) and terminal transverse limb defects, in association with variable involvement of the brain, eyes, and cardiovascular systems (summary by Shaheen et al., 2011). For a discussion of genetic heterogeneity of Adams-Oliver syndrome, see AOS1 (100300). Clinical Features Koiffmann et al. (1988) reported a Brazilian family with Adams-Oliver syndrome suggesting autosomal recessive inheritance. ... McGoey and Lacassie (2008) reported 2 sisters with Adams-Oliver syndrome who had central nervous system abnormalities. ... McGoey and Lacassie (2008) reviewed previous reports of autosomal recessive inheritance of Adams-Oliver syndrome, stating that 12 patients from 9 kindreds had been reported. ... Inheritance The transmission pattern of Adams-Oliver syndrome in the families reported by Shaheen et al. (2011) was consistent with autosomal recessive inheritance.
A rare disorder characterized by the combination of congenital limb abnormalities and scalp defects, often accompanied by skull ossification defects. Epidemiology The prevalence is unknown. Clinical description The severity of the disorder varies greatly among affected individuals. Aplasia cutis congenita, transverse limb defects and cutis marmorata telangiectica are characteristic of this condition. The affected patients typically have malformations of the hands, arms, feet and/or legs that range from hypoplastic fingers and toes to absent hands and/or lower legs, and occasionally show intellectual deficit. AOS may be associated with a variety of physical anomalies including congenital cataract, strabismus and microphthalmia, congenital heart malformations (including tetralogy of Fallot and pulmonary atresia), and hepatoportal sclerosis.
A number sign (#) is used with this entry because of evidence that Adams-Oliver syndrome-1 (AOS1) is caused by heterozygous mutation in the ARHGAP31 gene (610911) on chromosome 3q13. ... Among the 31 reported patients with the full syndrome, major hemorrhage from the scalp defect occurred in 10, with 2 fatalities. ... Zapata et al. (1995) reported 2 patients with Adams-Oliver syndrome and congenital cardiac malformations. A literature review demonstrated that 13.4% of individuals with this syndrome have congenital heart anomalies. ... The report was further evidence that the Adams-Oliver syndrome may be a vascular disruption sequence.
Most children with Adams–Oliver syndrome can likely expect to have a normal life span. ... "Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies" . ... "Haploinsufficiency of the NOTCH1 Receptor as a Cause of Adams-Oliver Syndrome With Variable Cardiac Anomalies" . ... Whitley CB, Gorlin RJ (1991). "Adams–Oliver syndrome revisited". Am J Med Genet . 40 (3): 319–326. doi : 10.1002/ajmg.1320400315 . ... "Congenital cardiac malformations in Adams–Oliver syndrome". Clin Genet . 47 (2): 80–84. doi : 10.1111/j.1399-0004.1995.tb03928.x .
Adams-Oliver syndrome (AOS) is a rare disease characterized by an abnormality of skin development (areas of missing skin on the scalp called aplasia cutis congenita ) and malformations of the hands and feet (terminal transverse limbs defects). ... Severity can vary greatly among people with the syndrome and may be lethal in some cases.
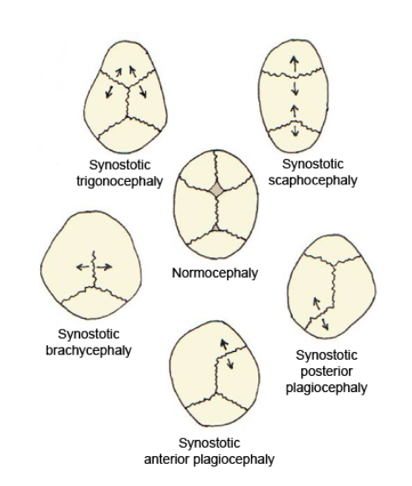
A number sign (#) is used with this entry because of evidence that susceptibility to craniosynostosis-5 (CRS5) is caused by variation in the ALX4 gene (605420) on chromosome 11p11. Description Premature fusion of the various sutures in the human neurocranium (skull vault and base) is defined as craniosynostosis (CRS). Clinical consequences include abnormal head shape and increased intracranial pressure, which may result in neurologic symptoms, developmental delay, and hearing or vision problems. Approximately 80% of cases are classified as nonsyndromic craniosynostosis and present as isolated suture fusion with no other associated anomalies. Sagittal suture fusion is the most common form of isolated craniosynostosis, accounting for 40 to 58% of all isolated cases (summary by Yagnik et al., 2012).
Differential diagnosis Sagittal synostosis can be isolated or occur as part of a syndrome (such as familial scaphocephaly with radioulnar synostosis or sagittal craniosynostosis with Dandy-Walker malformation and hydrocephalus; see these terms). The syndromic and nonsyndromic forms can be clearly distinguished due to the absence in isolated scaphocephaly of additional clinical features.
Rieubland et al. (2011) concluded that the patient represented a previously undescribed craniosynostosis syndrome. Twigg et al. (2013) studied 26 mutation-positive individuals from 12 families with craniosynostosis associated with heterozygous mutation in the ERF gene (see MOLECULAR GENETICS). ... In the 8 children who underwent 3D CT scan of the skull, fusion was observed to have affected the sagittal suture in 7 cases, the lambdoid suture in 5 cases, the coronal suture in 3 cases, and the metopic suture in 1 case. Seven of 12 probands had syndromic multiple suture synostosis, 2 had only sagittal synostosis, 1 had only lambdoid synostosis, and in 1 proband the sutures were not specified. In half of the families, a diagnosis of Crouzon syndrome (123500) had been suggested because of exorbitism and midface hypoplasia, but FGFR2 (176943) testing was normal.
Description Schimmelpenning-Feuerstein-Mims syndrome, also known as linear sebaceous nevus syndrome, is characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects (summary by Happle, 1991 and Ernst et al., 2007). ... Consistent with the proposal by Happle (1991), Dodge and Dobyns (1995) suggested that sebaceous nevus syndrome may be caused by mosaic mutation of a gene that would be lethal if expressed in all cells. ... Inheritance Schimmelpenning-Feuerstein-Mims syndrome is believed to be caused by an autosomal dominant lethal mutation that survives by somatic mosaicism. ... The report supported the concept of a postzygotic somatic mutation in the etiology of the syndrome. Rijntjes-Jacobs et al. (2010) reported a second case of monozygotic twins discordant for SFM syndrome. ... Happle (1986) suggested a similar mechanism for the Proteus syndrome (176920) and the McCune-Albright syndrome (174800).
Linear nevus sebaceous syndrome (LNSS) is a condition characterized by the association of a large, linear sebaceous nevus (type of birthmark) with a broad range of abnormalities that may affect every organ system, including the central nervous system (CNS).
Ischio-vertebral syndrome is a very rare, poorly-defined bone disease characterized by ischial aplasia or hypoplasia, vertebral anomalies (vertebral malsegmentation, kyphoscoliosis), and in some patients, non-distinctive facial dysmorphism.
Edwards et al. (1989) concluded that the condition is distinct from Alport syndrome (301050), which usually includes hematuria, is not associated with hyperparathyroidism, and is X-linked in its best studied form, although there is an autosomal dominant (104200) and possibly an autosomal recessive (203780) form.
A rare, genetic, syndromic intellectual disability characterized by intrauterine growth retardation, microcephaly, hypotonia, motor and neurodevelopmental delay, speech delay, intellectual disability, and mild dysmorphic features.
An aggressive soft tissue cancer that typically arises in serous lined surfaces of the abdominal or pelvic peritoneum, and spreads to the omentum, lymph nodes and hematogenously disseminates especially to the liver. Extraserous primary location has been reported in exceptional cases. Epidemiology DSRCT is extremely rare. Only a few hundred cases have been reported worldwide since the first description in 1989. It usually affects males, during adolescence or young adulthood, with a male-to-female ratio of 4:1. Clinical description Clinical signs and symptoms of DSRCT are non-specific.
Desmoplastic small round cell tumor (DSRCT) is a rare type of soft tissue cancer (sarcoma) that usually begins in the abdomen. It primarily affects children and young adults and is more common in males. It is formed by small, round cancer cells surrounded by scar-like tissue and is often found in the tissue (peritoneum) that lines the inside of the abdomen and pelvis. The tumor cells have a characteristic genetic change involving a translocation between chromosomes 11 and 22 , which is important in differentiating from other similar tumors. The genetic change involved in DSRCT is acquired throughout a person's lifetime and is not inherited.
In the family reported by Moynahan (1962), 2 brothers were affected. The alopecia consisted of a delay in the growth of hair. The father of the boys had been bald until age 2 and a maternal aunt until age 4. Mosavy (1975) observed 4 affected sibs and Pfeiffer and Volklein (1982) reported affected brother and sister. Wessel et al. (1987) reported 3 sibs with alopecia, seizures, and mental retardation. In a family derived from Bangladesh, van Haeringen et al. (1990) described a father and 3 children (of 7) with microcephaly, sparse hair, mild to moderate mental retardation, and, in the 2 affected boys, generalized seizures.
Walsh and Montplaisir (1982) described an unusual family in which the mother and 4 of 9 sibs had glaucoma with maximal intraocular pressure in the morning and heavy snoring. Two other surviving brothers had only sleep apnea with no glaucoma. In those who had recordings, episodes of sleep apnea tended to occur and to be more prolonged in rapid-eye-movement sleep.
A number sign (#) is used with this entry because of evidence that the melanoma-astrocytoma syndrome is caused by mutation in the CDKN2A gene (600160) on chromosome 9p21. ... In 20 of 25 of the cutaneous melanoma patients in this series and in additional first-degree relatives of 9 of 11 of the affected families, atypical melanocytic nevi had occurred as are found in the familial atypical multiple mole-melanoma syndrome (155600). Bahuau et al. (1997) also described a family with a cancer syndrome of CMM and nervous system tumors. ... These phenotypic features extended beyond the melanoma-astrocytoma syndrome seen with isolated deletion of CDKN2A. ... Molecular Genetics In a family with melanoma-astrocytoma syndrome, Tachibana et al. (2000) identified a heterozygous germline deletion of the p16/CDKN2A gene. ... Pasmant et al. (2007) suggested that ANRIL might be involved in melanoma-NST syndrome families and in melanoma-prone families with no identified p16/CDKN2A mutations as well as in somatic tumors.
Melanoma and neural system tumor syndrome is an extremely rare tumor association characterized by dual predisposition to melanoma and neural system tumors (typically astrocytoma; see this term). ... Genetic mutations or germline deletions are thought to underlie this cancer susceptibility syndrome. Genetic counseling Male-to-male transmission was reported in one family but inheritance patterns are not clearly characterized.
A number sign (#) is used with this entry because of evidence that Alazami syndrome (ALAZS) is caused by homozygous or compound heterozygous mutation in the LARP7 gene (612026), a chaperone of 7SK noncoding RNA (616505), on chromosome 4q25. Description Alazami syndrome is an autosomal recessive disorder characterized by severe growth restriction present at birth, severely impaired intellectual development, and distinctive facial features. ... Imbert-Bouteille et al. (2019) reported 2 sisters, born to consanguineous Algerian parents, with Alazami syndrome. The authors reviewed the clinical information on 15 previously reported patients in addition to their 2 newly reported patients and confirmed the key features of the syndrome: severe growth restriction, severely impaired intellectual development, and distinguishing facial features, including broad nose, malar hypoplasia, wide mouth, full lips, and widely spaced teeth. ... In a 2-year-old Caucasian girl, born of unrelated parents, with Alazami syndrome, Ling and Sorrentino (2016) identified compound heterozygous frameshift mutations in the LARP7 gene (612026.0003 and 612026.0004). ... In 2 sisters, born to consanguineous Algerian parents, with Alazami syndrome, Imbert-Bouteille et al. (2019) identified a homozygous frameshift mutation in the LARP7 gene (612026.0005).
Epidemiology To date, less than 30 affected individuals reported worldwide, about half of which belong to a few consanguineous families. Clinical description Alazami syndrome is a genetic developmental defect characterized by mild to severe short stature, head circumference below the 50th centile, microcephaly in half of the individuals, and global developmental delay with moderate to severe intellectual disability. ... Features more rarely reported include cleft palate, brachydactyly, prominent interphalangeal joints, 2-3 toe syndactyly, metaphyseal dysplasia, hydronephrosis due ureteropelvic junction stenosis, and hypospadias. Etiology The syndrome is caused by biallelic loss-of-function variants in the LARP7>/i> gene (4q25), which encodes a protein involved in the regulation of RNA transcription and splicing. ... Differential diagnosis Differential diagnosis includes other syndromes characterized by intellectual disability and short stature. Antenatal diagnosis Prenatal diagnosis is available for at-risk pregnancies, if a pathogenic variant has been identified in a member of the affected family. Genetic counseling Alazami syndrome is an autosomal recessive disorder, with homozygous and compound heterozygous variants described.
Tubular renal disease-cardiomyopathy syndrome is characterised by hypokalaemic metabolic alkalosis secondary to a tubulopathy, hypomagnesaemia with hypermagnesuria, severe hypercalciuria and dilated cardiomyopathy.
A number sign (#) is used with this entry because of evidence that Stuve-Wiedemann syndrome (STWS), also known as neonatal Schwartz-Jampel syndrome type 2 (SJS2), is caused by mutation in the leukemia inhibitory factor receptor gene (LIFR; 151443) on chromosome 5p13. ... Superti-Furga et al. (1998) also concluded that Schwartz-Jampel syndrome type 2 and Stuve-Wiedemann syndrome should be considered the same entity. ... Patients with Longer Survival Stuve-Wiedemann syndrome is typically lethal in the neonatal period. ... This report confirmed that survival in this syndrome is possible and that the prognosis improves after the first year of life. ... Mapping Giedion et al. (1997) and Brown et al. (1997) demonstrated that neonatal Schwartz-Jampel syndrome type 2 was not linked to chromosome 1p36.1-p34, where 'classic' Schwartz-Jampel syndrome maps.
Goodman et al. (1987) called attention to the fact that the ICE acronym is used for the iridocorneal endothelial syndrome (Yanoff, 1979), a distinct disorder. Begas and Delleman (1988) begged that authors find another, less confusing name for the new syndrome. Limbs - Pes planus Facies - Prominent and full cheeks - Sparse lateral eyebrows Inheritance - Autosomal dominant Spine - Kyphoscoliosis Mouth - High-arched palate Skin - Ichthyosis vulgaris Thorax - Anterior chest deformity ▲ Close
Short stature-auditory canal atresia-mandibular hypoplasia-skeletal anomalies syndrome is a rare, genetic, multiple congenital anomalies/dysmorphic syndrome characterized by short stature, conductive hearing loss due to bilateral auditory canal atresia, mandibular hypoplasia and multiple skeletal abnormalities, including bilateral humeral hypoplasia, humeroscapular synostosis, delayed pubis rami ossification, central dislocation of the hips, and proximal femora defects, as well as bilateral talipes equinovarus, proximally implanted thumbs and lumbar hyperlordosis.
Description Short stature, auditory canal atresia, mandibular hypoplasia, and skeletal abnormalities (SAMS) is an autosomal recessive multiple congenital anomaly syndrome with features of a first and second branchial arch syndrome. ... Affected individuals may also have some features of a neurocristopathy or abnormal mesoderm development, such as urogenital anomalies, that are distinct from other branchial arch syndromes (summary by Parry et al., 2013). Clinical Features Lemire et al. (1998) described a 9-year-old girl, the daughter of consanguineous Mennonite parents, who had a seemingly unique multiple congenital anomaly syndrome consisting of short stature, auditory canal atresia, mandibular hypoplasia, and skeletal abnormalities.
17p13.3 microduplication syndrome is characterized by variable psychomotor delay and dysmorphic features. ... It encompasses the same region that is deleted in Miller-Dieker (17p13 deletion) syndrome (see this term).. The variable size of this de novo duplication indicates that mechanisms other than nonallelic homologous recombination (NAHR) may be responsible.
A number sign (#) is used with this entry because it represents a contiguous gene duplication syndrome involving the PAFAH1B1 (LIS1; 601545) and/or the YWHAE (605066) gene on chromosome 17p13.3. The same region on chromosome 17p13.3 is deleted in Miller-Dieker lissencephaly syndrome (MDLS; 247200). Clinical Features Bi et al. (2009) reported 7 unrelated individuals with different submicroscopic duplications of chromosome 17p13.3 involving the LIS1 and/or YWHAE genes.
Rowley et al. (1961) described a 'new' syndrome in 3 of 6 children. Features were growth retardation, poor muscular development, scanty adipose tissue, recurrent pulmonary infection, atelectasis, and right ventricular hypertrophy. ... The disorder is sometimes referred to as the 'Busby syndrome,' for the surname of the affected family.