An extremely rare genetic syndrome characterized by the association of microcephaly, intellectual deficit and achalasia (with symptoms of coughing, dysphagia, vomiting, failure to thrive and aspiration appearing in infancy/early-childhood).
Frequency 9 children between 1980-2017 Achalasia microcephaly syndrome is a rare condition whereby achalasia in the oesophagus manifests alongside microcephaly and mental retardation. This is a rare constellation of symptoms with a predicted familial trend. [1] The main signs of achalasia microcephaly syndrome involve the manifestation of each individual disease associated with the condition. ... The main symptoms of achalasia microcephaly syndrome are the progressive manifestation of the major symptoms associated with the individual diseases, in young children. ... "Another case of achalasia-microcephaly syndrome". Clinical Dysmorphology . 8 (4): 295–7. doi : 10.1097/00019605-199910000-00012 . ... S2CID 2186970 . ^ Williams JJ, Sandlin CS, Dumars KW (1978). "New syndrome: microcephaly associated with achalasia".
Clinical Features Williams et al. (1978) described 3 sisters and a brother with microcephaly, mental retardation, and early onset of symptoms of achalasia. The brother, who died in Mexico at age 4.5 years, had recurrent vomiting (Dumars et al., 1980). The parents denied consanguinity but came from the same small village in Mexico. Hernandez et al. (1989) stated that 2 of the affected sibs in this family had initially been reported by Polonsky and Guth (1970) as having familial achalasia (200400). Khalifa (1988) reported 2 Libyan brothers, aged 7 and 9 years, with achalasia of the cardia, microcephaly, and mental retardation.
A number sign (#) is used with this entry because of evidence that RHYNS syndrome is caused by compound heterozygous mutation in the TMEM67 gene (609884) on chromosome 8q22. One such patient has been reported. Description RHYNS syndrome is characterized by gaze palsy, retinitis pigmentosa, sensorineural hearing loss, hypopituitarism, nephronophthisis, and mild skeletal dysplasia (Di Rocco et al., 1997). ... Di Rocco et al. (1997) designated the syndrome 'RHYNS,' for retinitis pigmentosa, hypopituitarism, nephronophthisis, and skeletal dysplasia. ... The authors suggested that the phenotype was consistent with RHYNS syndrome. Brancati et al. (2018) restudied the Italian patient with RHYNS syndrome who was originally reported by Di Rocco et al. (1997). ... Inheritance The presence of RHYNS syndrome in 2 brothers described by Hedera and Gorski (2001) supported an autosomal recessive mode of inheritance; however, the authors noted that all 4 reported cases were male, and thus an X-linked mode of inheritance could not be excluded.
Distal arthrogryposis type 4 is an inherited developmental defect syndrome characterized by multiple congenital contractures of limbs, without primary neurologic and/or muscle disease that affects limb function, and a mild to severe scoliosis.
The proband's uncle had features similar to those of the proband's father, and photos of a paternal grandfather suggested that he also had the same syndrome. Nomenclature In a revised and extended classification scheme of the distal arthrogryposes, Bamshad et al. (1996) referred to this disorder as distal arthrogryposis type 4 (DA4).
Rare non-syndromic intellectual disability is a rare, hereditary, neurologic disease characterized by early-onset cognitive impairment as a sole disability.
Syndromic orbital border hypoplasia is a rare disorder observed in two families to date and characterized by agenesis of the orbital margin, varying defects of the lacrimal passages, hypoplasia of the palpebral skin and tarsal plates and atresia of the nasolacrimal duct.
Urrets-Zavalia (1955) observed 2 families with a syndrome consisting of agenesis of the orbital margin, hypoplasia of the palpebral skin and tarsal plates, and variable defects of the lacrimal passages including ectopia and elongation of the lower punctum, shortening or absence of the inferior canaliculi, supernumerary canaliculi, or atresia of the nasolacrimal duct.
This syndrome is characterized by the association of hypogonadotropic hypogonadism (with primary amenorrhea and lack of secondary sexual development) and retinitis pigmentosa (see this term).
Normal steroid sulfatase excluded the X-linked form (308100). X-linked inheritance of the syndrome is, of course, possible. The hepatosplenomegaly suggested a storage disease, but its nature was not evident.
3q26 microduplication syndrome is a rare chromosomal anomaly characterized by prenatal and postnatal growth retardation, developmental delay, intellectual impairment, dysmorphic signs and variable combination of congenital anomalies, including cardiovascular, genitourinary and skeletal anomalies and spectrum of caudal malformations.
Combined presence of Wernicke's encephalopathy (WE) and Korsakoff's syndrome Wernicke–Korsakoff syndrome Other names Korsakoff's psychosis, alcoholic encephalopathy, [1] "wet brain" [2] Thiamine Specialty Psychiatry , neurology Wernicke–Korsakoff syndrome ( WKS ) is the combined presence of Wernicke encephalopathy (WE) and alcoholic Korsakoff syndrome . ... Later, it was found that Wernicke encephalopathy and alcoholic Korsakoff syndrome are products of the same cause. [36] Alcoholic Korsakoff syndrome [ edit ] Sergei Korsakoff was a Russian physician after whom the disease "Korsakoff's syndrome" was named. ... After the presentation of this report the term "Korsakoff's syndrome" was coined. [25] Although WE and AKS were discovered separately, these two syndromes are usually referred to under one name, Wernicke–Korsakoff syndrome, due to the fact that they are part of the same cause and because the onset of AKS usually follows WE if left untreated. [ citation needed ] Society and culture [ edit ] The British neurologist Oliver Sacks describes case histories of some of his patients with the syndrome in the book The Man Who Mistook His Wife for a Hat (1985). ... D.; Guerrini, I.; Marshall, E. J. (2009). "The Korsakoff Syndrome: Clinical Aspects, Psychology and Treatment" . ... "Development of Wernicke-Korsakoff syndrome after long intervals following gastrectomy" .
Find sources: "Mare reproductive loss syndrome" – news · newspapers · books · scholar · JSTOR ( February 2014 ) ( Learn how and when to remove this template message ) Mare reproductive loss syndrome ( MRLS ) is a syndrome consisting of equine abortions and three related nonreproductive syndromes which occur in horses of all breeds, sexes, and ages. ... Based on these overwhelming reproductive losses, the syndrome was named the mare reproductive loss syndrome (MRLS). MRLS was defined as including four syndromes: (1) EFLs, (2) LFLs, (3) unique unilateral uveitis, and (4) pericarditis syndrome. An associated encephalitis syndrome was not included in the original case definition. ... Clinical signs [ edit ] MRLS was initially characterized by four syndromes: (1) EFLs, (2) LFLs and the nonreproductive syndromes, (3) unilateral uveitis, (4) pericarditis, and later (5) Actinobacillus encephalitis .
Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. ... Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. ... Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. ... Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. ... Epilepsia 2007; 48(6):1165-1172. ^ Panayiotopoulos CP. Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome.
Benign childhood occipital epilepsy, Panayiotopoulos type is a rare, genetic neurological disorder characterized by late infancy to early-adolescence onset of prolonged, nocturnal seizures which begin with autonomic features (e.g. vomiting, pallor, sweating) and associate tonic eye deviation, impairment of consciousness and may evolve to a hemi-clonic or generalized convulsion. Autonomic status epilepticus may be the only clinical event in some cases.
Gaucher disease - ophthalmoplegia - cardiovascular calcification is a variant of Gaucher disease, also known as a Gaucher-like disease that is characterized by cardiac involvement. Epidemiology This syndrome is rare with less than 30 cases reported in the literature. ... Antenatal diagnosis Prenatal diagnosis is possible through detection of the glucosylceramidase deficiency in amniocytes or chorionic villus samples, or through screening for the GBA gene mutation in families in which the D409H allele has been identified in both parents or an affected brother or sister. Genetic counseling The syndrome is transmitted in an autosomal recessive manner. Management and treatment Patients with this syndrome require close monitoring by echocardiography as the cardiac complications require aortic and mitral valve replacement.
Clinical Features Verloes et al. (1989) reported a brother and sister and probably a third sib with a seemingly characteristic and previously undescribed syndrome. Microcephaly was severe and there was also microphthalmia, brachydactyly with clinodactyly 5, delayed growth in puberty, and severe mental retardation. ... Bottani and Verloes (1995) suggested that the disorder they described might be the same as the growth-mental deficiency syndrome of Myhre (139210). Farrell (1997) described a 23-year-old male with microphthalmia, severe developmental delay, conductive hearing loss, marked short stature of prenatal onset, and radiographic skeletal changes. ... Cytogenetics Verloes et al. (2000) used subtelomeric probes to reinvestigate the family initially published as GOMBO syndrome by Verloes et al. (1989). They identified a cryptic translocation resulting in 3p monosomy and 22q trisomy.
Moyamoya angiopathy - short stature - facial dysmorphism - hypergonadotropic hypogonadism is a very rare, hereditary, neurological, dysmorphic syndrome characterized by moyamoya disease, short stature of postnatal onset, and stereotyped facial dysmorphism. Epidemiology The syndrome is extremely rare and has been reported in three unrelated families to date, with 10 affected individuals in several generations. ... Genetic counseling Reported cases are suggestive of a hereditary syndrome with an X-linked recessive pattern of inheritance.
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome on chromosome Xq28. Description This multisystem disorder is characterized by moyamoya disease, short stature, hypergonadotropic hypogonadism, and facial dysmorphism. ... Inheritance The transmission pattern of syndromic moyamoya disease in the families reported by Herve et al. (2010) and Miskinyte et al. (2011) was consistent with X-linked recessive inheritance. Molecular Genetics In affected members of 3 families with X-linked recessive syndromic moyamoya disease, Miskinyte et al. (2011) identified 3 different deletions on chromosome Xq28. ... Miskinyte et al. (2011) noted that some of the features of the disorder were reminiscent of chromosome breakage syndromes. INHERITANCE - X-linked recessive GROWTH Height - Short stature HEAD & NECK Face - Long philtrum - Retrognathia Ears - Low-set ears Eyes - Early-onset cataracts (1 family) - Ptosis - Hypertelorism - Deep-set eyes Nose - Wide nose - Broad nose - Flared nares CARDIOVASCULAR Heart - Dilated cardiomyopathy (variable) - Left ventricular enlargement (variable) Vascular - Moyamoya disease - Cerebrovascular disease GENITOURINARY External Genitalia (Male) - Decreased testicular volume Internal Genitalia (Male) - Azoospermia SKELETAL Hands - Small hands - Short, broad fingers SKIN, NAILS, & HAIR Hair - Premature graying NEUROLOGIC Central Nervous System - Acute neurologic deficits due to cerebrovascular disease - Cerebral infarcts - Cerebral hemorrhages - Stroke-like symptoms - Seizures (less common) - Developmental delay (1 family) ENDOCRINE FEATURES - Hypergonadotropic hypogonadism - Growth hormone deficiency MISCELLANEOUS - Three families have been reported (as of 28 June 2011) - Onset of neurologic events can occur between 4 and 35 years of age - Facial dysmorphic features are variable MOLECULAR BASIS - Contiguous gene deletion syndrome caused by deletion (3.4 kb) of chromosome Xq28 ▲ Close
Tethered cord syndrome is a rare neurological condition. ... Infants and children with congenital tethered cord syndrome may also have tufts of hair, dimples, skin discoloration, or benign fatty tumors on the lower back. Tethered cord syndrome occurs when tissue attachments limit the movement of the spinal cord within the spinal column.
ISBN 978-0-8089-2366-4 . v t e Lupus nephritis Class I ( Minimal mesangial glomerulonephritis ) Class II ( Mesangial proliferative lupus nephritis ) Class III ( Focal proliferative nephritis ) Class IV ( Diffuse proliferative nephritis ) Class V ( Membranous nephritis ) Class VI ( Glomerulosclerosis ) v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy This article about a disease of the genitourinary system is a stub .
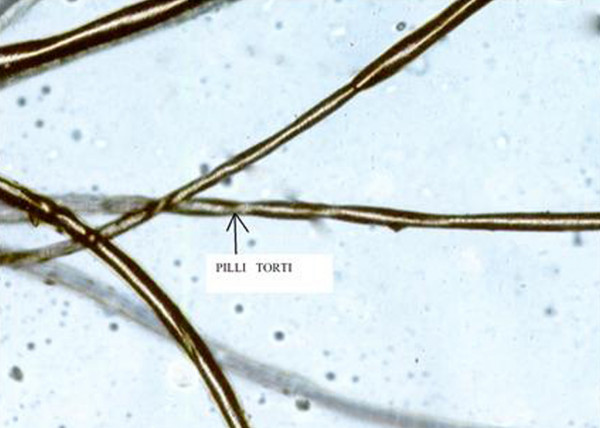
Pili torti Menkes disease Specialty Medical genetics Pili torti (also known as "Twisted hairs") is characterized by short and brittle hairs that appear flattened and twisted when viewed through a microscope. [1] : 638 [2] : 764 [3] This phenotype is noted in Menkes disease , and Lichen Planopilaris Pili torti can also occur after use of retinoids, such as isotretinoin See also [ edit ] List of cutaneous conditions Crandall syndrome References [ edit ] ^ Freedberg, et al. (2003). ... External links [ edit ] Classification D ICD - 10 : Q84.1 ( ILDS Q84.120) MeSH : C562485 C562485, C562485 DiseasesDB : 29682 v t e Congenital malformations and deformations of skin appendages Nail disease Anonychia Leukonychia Pachyonychia congenita / Onychauxis Koilonychia Hair disease hypotrichosis /abnormalities: keratin disease Monilethrix IBIDS syndrome Sabinas brittle hair syndrome Pili annulati Pili torti Uncombable hair syndrome Björnstad syndrome Giant axonal neuropathy with curly hair hypertrichosis : Zimmermann–Laband syndrome This condition of the skin appendages article is a stub .
Pili torti is also a feature in several disorders, including Bjornstad syndrome (BJS; 262000), Bazex syndrome (BZX; 301845), and Menkes disease (309400).
It can occur alone or as part of other diseases like ectodermal dysplasias , Menke disease , Bjornstand syndrome , or Bazex syndrome . Acquired cases of pili torti may be associated with anorexia nervosa , malnutrition, oral retinoid treatment, or inflammatory scalp conditions (e.g., cutaneous lupus erythematousus ).
Myelodysplastic syndrome with excess blasts is a rare type of myelodysplastic syndrome (MDS). ... Some cases of MDS are linked to known risk factors (such as smoking, chemotherapy, having a genetic syndrome that increases the chance of developing MDS and other).
A rare hematologic disease characterized by the presence of 20-29% blasts in the bone marrow, presence of 5-29% blasts in the peripheral blood, and/or presence of Auer rods. Patients show relatively stable peripheral blood counts for weeks or months, with specific cytogenetic and molecular genetic characteristics constituting important prognostic factors.
Refractory anemia' with excess of blasts Specialty Oncology Refractory anemia with excess of blasts (RAEB) is a type of myelodysplastic syndrome [1] with a marrow blast percentage of 5% to 19%. [2] In MeSH , "Smoldering leukemia" is classified under RAEB. [3] References [ edit ] ^ Palmieri S, D'Arco AM, Celentano M, et al. ... National Cancer Institute document: "Dictionary of Cancer Terms" . v t e Myeloid -related hematological malignancy CFU-GM / and other granulocytes CFU-GM Myelocyte AML : Acute myeloblastic leukemia M0 M1 M2 APL/M3 MP Chronic neutrophilic leukemia Monocyte AML AMoL/M5 Myeloid dendritic cell leukemia CML Philadelphia chromosome Accelerated phase chronic myelogenous leukemia Myelomonocyte AML M4 MD-MP Juvenile myelomonocytic leukemia Chronic myelomonocytic leukemia Other Histiocytosis CFU-Baso AML Acute basophilic CFU-Eos AML Acute eosinophilic MP Chronic eosinophilic leukemia / Hypereosinophilic syndrome MEP CFU-Meg MP Essential thrombocytosis Acute megakaryoblastic leukemia CFU-E AML Erythroleukemia/M6 MP Polycythemia vera MD Refractory anemia Refractory anemia with excess of blasts Chromosome 5q deletion syndrome Sideroblastic anemia Paroxysmal nocturnal hemoglobinuria Refractory cytopenia with multilineage dysplasia CFU-Mast Mastocytoma Mast cell leukemia Mast cell sarcoma Systemic mastocytosis Mastocytosis : Diffuse cutaneous mastocytosis Erythrodermic mastocytosis Adult type of generalized eruption of cutaneous mastocytosis Urticaria pigmentosa Mast cell sarcoma Solitary mastocytoma Systemic mastocytosis Xanthelasmoidal mastocytosis Multiple/unknown AML Acute panmyelosis with myelofibrosis Myeloid sarcoma MP Myelofibrosis Acute biphenotypic leukaemia This oncology article is a stub .
Refractory anemia with excess blasts (RAEB) is a frequent severe subtype of myelodysplastic syndrome (MDS; see this term) characterized by cytopenias with unilineage or multilineage dysplasia and 5% to 19% blasts in bone marrow or blood.
A very rare subtype of Waardenburg syndrome (WS) that is characterized by limb anomalies in association with congenital hearing loss, minor defects in structures arising from neural crest, resulting in pigmentation anomalies of eyes, hair, and skin. Epidemiology Incidence is unknown, but WS3 is the rarest form of all Waardenburg syndrome types. Clinical description WS3 is characterized by the association of limb anomalies (predominantly involving upper limbs, with hypoplasia of the musculoskeletal system, flexion contractures, fusion of the carpal bones, syndactylia) with features of Waardenburg syndrome (see this term), which include congenital sensorineural hearing loss, hypopigmentation abnormalities of irides, hair and skin and minor facial dysmorphism in combination with dystopia canthorum.