-
Microtia-Anotia
Omim
For instance, M-A is an essential component of isotretinoin embryopathy (243440), is an important manifestation of thalidomide embryopathy, and can be part of the prenatal alcohol syndrome and maternal diabetes embryopathy. M-A occurs with a number of single gene disorders, such as Treacher Collins syndrome (154500), branchiotorenal/branchiootic syndromes (see 113650 and 602588), oculoauricular syndrome (612109), microtia with hearing impairment and cleft palate (612290), or chromosomal syndromes, such as trisomy 18. M-A also occurs as part of seemingly nonrandom patterns of multiple defects, such as Goldenhar syndrome (164210) (Mastroiacovo et al., 1995). Alasti and Van Camp (2009) reviewed the genetics of microtia and microtia-associated syndromes and discussed their clinical aspects in relation to the causative genes. ... Of the 172 infants, 114 (66.2%) had an isolated defect, 48 (27.9%) were multimalformed infants (MMI) with M-A, and 10 (5.8%) had a well defined syndrome. The frequency of bilateral defects among nonsyndromic cases was 12% compared to 50% of syndromic cases.

-
Ruijs-Aalfs Syndrome
Wikipedia
Unsourced material may be challenged and removed. Find sources: "Ruijs-Aalfs syndrome" – news · newspapers · books · scholar · JSTOR ( June 2019 ) ( Learn how and when to remove this template message ) This article includes a list of general references , but it remains largely unverified because it lacks sufficient corresponding inline citations . ... Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( June 2019 ) ( Learn how and when to remove this template message ) Ruijs-Aalfs syndrome Autosomal recessive pattern is the inheritance manner of this condition Specialty Medical genetics Causes Mutations in the SPRTN gene Ruijs-Aalfs syndrome is a rare condition characterised by facial and skeletal abnormalities along with the development of hepatoma in the teenage years. ... Spartan is intimately involved in the repair of protein-linked DNA breaks. [2] Pathopysiology [ edit ] This is not understood. Diagnosis [ edit ] This syndrome may be suspected on clinical grounds. The diagnosis is established by sequencing the SPRTN gene Differential diagnosis [ edit ] Werner syndrome Treatment [ edit ] There is no specific treatment for this condition. ... PMID 31331820 ^ Ruijs MWG, van Andel RNJ, Oshima J, Madan K, Nieuwint AWM, Aalfs CM (2003) Atypical progeroid syndrome: an unknown helicase gene defect?

-
Pitt-Hopkins-Like Syndrome 2
Omim
A number sign (#) is used with this entry because Pitt-Hopkins-like syndrome-2 (PTHSL2) is caused by compound heterozygous mutation in the NRXN1 gene (600565) on chromosome 2p16. Clinical Features Zweier et al. (2009) reported an 18-year-old girl with a mental retardation syndrome resembling Pitt-Hopkins syndrome (PTHS; 610954). ... Harrison et al. (2011) reported 2 sisters, born of unrelated Caucasian parents, with a severe mental retardation syndrome characterized by onset of epileptic encephalopathy at age 4 and 6 months, respectively, with loss of early developmental skills. ... Molecular Genetics In a girl with Pitt-Hopkins-like syndrome-2, Zweier et al. (2009) identified compound heterozygosity for 2 mutations in the NRXN1 gene (600565.0001 and 600565.0002). In 2 sisters with a severe early-onset mental retardation syndrome with severe epilepsy, Harrison et al. (2011) identified compound heterozygous deletions on chromosome 2p16.3, exclusively affecting the NRXN1 gene (600565.0004 and 600565.0005).
-
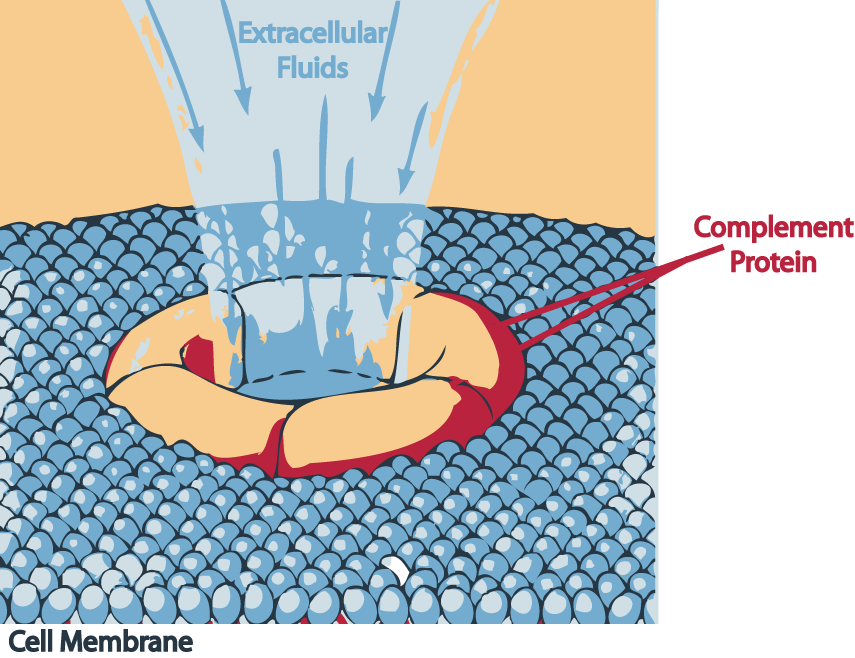
Terminal Complement Pathway Deficiency
Wikipedia
External links [ edit ] Classification D OMIM : 120950 609536 , 612446 , 610102 v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency This genetic disorder article is a stub .
-
Premature Junctional Contraction
Wikipedia
University of Utah. v t e Cardiovascular disease (heart) Ischaemic Coronary disease Coronary artery disease (CAD) Coronary artery aneurysm Spontaneous coronary artery dissection (SCAD) Coronary thrombosis Coronary vasospasm Myocardial bridge Active ischemia Angina pectoris Prinzmetal's angina Stable angina Acute coronary syndrome Myocardial infarction Unstable angina Sequelae hours Hibernating myocardium Myocardial stunning days Myocardial rupture weeks Aneurysm of heart / Ventricular aneurysm Dressler syndrome Layers Pericardium Pericarditis Acute Chronic / Constrictive Pericardial effusion Cardiac tamponade Hemopericardium Myocardium Myocarditis Chagas disease Cardiomyopathy Dilated Alcoholic Hypertrophic Tachycardia-induced Restrictive Loeffler endocarditis Cardiac amyloidosis Endocardial fibroelastosis Arrhythmogenic right ventricular dysplasia Endocardium / valves Endocarditis infective endocarditis Subacute bacterial endocarditis non-infective endocarditis Libman–Sacks endocarditis Nonbacterial thrombotic endocarditis Valves mitral regurgitation prolapse stenosis aortic stenosis insufficiency tricuspid stenosis insufficiency pulmonary stenosis insufficiency Conduction / arrhythmia Bradycardia Sinus bradycardia Sick sinus syndrome Heart block : Sinoatrial AV 1° 2° 3° Intraventricular Bundle branch block Right Left Left anterior fascicle Left posterior fascicle Bifascicular Trifascicular Adams–Stokes syndrome Tachycardia ( paroxysmal and sinus ) Supraventricular Atrial Multifocal Junctional AV nodal reentrant Junctional ectopic Ventricular Accelerated idioventricular rhythm Catecholaminergic polymorphic Torsades de pointes Premature contraction Atrial Junctional Ventricular Pre-excitation syndrome Lown–Ganong–Levine Wolff–Parkinson–White Flutter / fibrillation Atrial flutter Ventricular flutter Atrial fibrillation Familial Ventricular fibrillation Pacemaker Ectopic pacemaker / Ectopic beat Multifocal atrial tachycardia Pacemaker syndrome Parasystole Wandering atrial pacemaker Long QT syndrome Andersen–Tawil Jervell and Lange-Nielsen Romano–Ward Cardiac arrest Sudden cardiac death Asystole Pulseless electrical activity Sinoatrial arrest Other / ungrouped hexaxial reference system Right axis deviation Left axis deviation QT Short QT syndrome T T wave alternans ST Osborn wave ST elevation ST depression Strain pattern Cardiomegaly Ventricular hypertrophy Left Right / Cor pulmonale Atrial enlargement Left Right Athletic heart syndrome Other Cardiac fibrosis Heart failure Diastolic heart failure Cardiac asthma Rheumatic fever
-
Cannabinoid Hyperemesis Syndrome
Wikipedia
"Cases of Death Secondary to Cannabinoid Hyperemesis Syndrome: 2217" . American Journal of Gastroenterology . pp. S1063. ^ a b Lu, ML; Agito, MD (July 2015). "Cannabinoid hyperemesis syndrome: Marijuana is both antiemetic and proemetic". ... (October 2019). "Cannabinoid hyperemesis syndrome" . Nursing . 49 (10): 42–45. doi : 10.1097/01.NURSE.0000577992.82047.67 . ... "Successful Treatment of Cannabinoid Hyperemesis Syndrome with Topical Capsaicin" . ACG Case Reports Journal . 5 : e3. doi : 10.14309/crj.2018.3 . ... "The Prevalence of Cannabinoid Hyperemesis Syndrome Among Regular Marijuana Smokers in an Urban Public Hospital" .

-
Aymé-Gripp Syndrome
Gene_reviews
Each child of an individual with Aymé-Gripp syndrome has a 50% chance of inheriting the MAF pathogenic variant. ... Formal clinical diagnostic criteria for Aymé-Gripp syndrome have not been established. Suggestive Findings Aymé-Gripp syndrome should be suspected in individuals with the following major and minor clinical features and imaging findings. ... For an in-depth analysis of the skeletal features seen in Aymé-Gripp syndrome, see the review by Niceta et al [2020]. ... Stickler syndrome caused by pathogenic variants COL2A1 , COL11A1 , or COL11A2 is inherited in an autosomal dominant manner; Stickler syndrome caused by pathogenic variants in COL9A1 , COL9A2 , or COL9A3 is inherited in an autosomal recessive manner. 2. Mild-to-moderate intellectual disability and developmental delay are common in Myhre syndrome; however, cognition can be within the normal range. 3.
- Musculocontractural Ehlers-Danlos Syndrome Gard
-
Syndactyly-Nystagmus Syndrome Due To 2q31.1 Microduplication
Orphanet
A rare, genetic, chromosomal anomaly syndrome resulting from partial duplication of the long arm of chromosome 2 characterized by congenital pendular nystagmus associated with bilateral cutaneous syndactyly between the third and fourth fingers.
-
Hemorrhagic Fever-Renal Syndrome
Orphanet
Hemorrhagic fever with renal syndrome (HFRS) is a rodent-borne potentially severe hemorrhagic disease caused by Old World Hantaviruses characterized by high fever, malaise, headache, myalgia, arthralgia, backache, abdominal pain, oliguria/renal failure and systemic hemorrhagic manifestations.
-
Ring Chromosome 9 Syndrome
Orphanet
Ring chromosome 9 syndrome is an autosomal anomaly characterized by variable clinical features, most commonly including developmental delay, some degree of intellectual disability, facial dysmorphism, microcephaly, congenital heart anomalies, and variable genital, limb and skeletal anomalies.
- Seizures-Intellectual Disability Due To Hydroxylysinuria Syndrome Orphanet
-
Ptosis-Vocal Cord Paralysis Syndrome
Orphanet
Ptosis-vocal cord paralysis syndrome is a rare, hereditary disorder with ptosis characterized by the combination of congenital bilateral recurrent laryngeal nerve paralysis and congenital bilateral ptosis.
-
Fibular Dimelia-Diplopodia Syndrome
Orphanet
A very rare, genetic, congenital limb malformation syndrome characterized by duplication of the fibula associated with pre-axial mirror polydactyly of the foot, that may occur as an isolated malformation or be assoicated with other anomalies, including ulnar dimelia, facial abnormalities and sacrococcygeal teratoma.
-
Aniridia-Absent Patella Syndrome
Orphanet
A rare syndrome described in three members of a family (a boy, his father, and his paternal grandmother) that is characterized by the association of aniridia with patella aplasia or hypoplasia.
-
Otoonychoperoneal Syndrome
Orphanet
A rare multiple congenital anomalies/dysmorphic syndrome characterized by the association of dysplastic external ears, nail hypoplasia, and variable skeletal malformations, such as hypoplastic or absent fibulae, abnormalities of the scapula, clavicle, and acromioclavicular joint, and talipes equinovarus, among others.
-
Lown-Ganong-Levine Syndrome
Orphanet
Lown-Ganong-Levine syndrome is an extremely rare conduction disorder characterized by a short PR interval (less than or equal to 120 ms) with normal QRS complex on electrocardiogram associated with the occurrence of episodes of atrial tachyarrythmias (e.g. atrial fibrillation, atrial tachycardia).
-
Olivopontocerebellar Atrophy-Deafness Syndrome
Orphanet
Olivopontocerebellar atrophy-deafness syndrome is characterised by infancy-onset olivopontocerebellar atrophy, sensorineural deafness and speech impairment.
-
Jung Syndrome
Orphanet
A rare, congenital malformation syndrome characterized by the association of anterior ocular chamber cleavage disorder with developmental delay, short stature and congenital hypothyroidism.
-
Diffuse Palmoplantar Keratoderma-Acrocyanosis Syndrome
Orphanet
Diffuse palmoplantar keratoderma-acrocyanosis syndrome is characterised by the association of diffuse palmoplantar keratoderma and acrocyanosis.



