-
Congenital Short Bowel Syndrome
Orphanet
Congenital short bowel syndrome is a rare intestinal disorder of neonates of unknown etiology.
-
Spastic Tetraplegia-Retinitis Pigmentosa-Intellectual Disability Syndrome
Orphanet
A rare, genetic, syndromic intellectual disability disorder characterized by the association of nonprogressive spastic quadriparesis, retinitis pigmentosa, intellectual disability, and variable deafness.
-
Irf6-Related Disorders
Gene_reviews
IRF6 -related disorders span a spectrum from isolated cleft lip and palate and Van der Woude syndrome (VWS) at the mild end to popliteal pterygium syndrome (PPS) at the more severe end. ... Presence of psychomotor disabilities may be seen in rare individuals with Van der Woude syndrome or popliteal pterygium syndrome. ... This type of mixed clefting can also occur with mutation of MSX1 [van den Boogaard et al 2000], TP63 , and FGFR1 and can be seen in individuals with 22q11.2 deletion syndrome, fetal alcohol syndrome, Kabuki syndrome, and CHARGE syndrome (see Differential Diagnosis). ... The mixed clefting seen in IFR6 -related disorders can also occur in MSX1 -related disorders [van den Boogaard et al 2000], TP63 -related disorders (e.g., ankyloblepharon-ectodermal defects-cleft lip/palate syndrome), FGFR1 -related disorders (e.g., isolated gonadotropin-releasing hormone deficiency), 22q11.2 deletion syndrome, fetal alcohol syndrome [Shaw & Lammer 1999], and CHARGE syndrome. ... These may also be seen in ankyloblepharon-ectodermal defects-cleft lip/palate syndrome, Rapp-Hodgkin syndrome (OMIM 129400), ectrodactyly, ectodermal dysplasia, cleft lip/palate syndrome 1 (OMIM 129900), curly hair-ankyloblepharon-nail dysplasia syndrome (OMIM 214350), and trisomy 18.
-
Spondylodysplastic Ehlers-Danlos Syndrome
Gard
Spondylodysplastic Ehlers-Danlos syndrome (EDS) is a subtype of the EDS, a group of genetic disorders of the connective tissue, which is the material between body cells that gives tissues form and strength. Ehlers-Danlos syndromes primarily affects the skin, hair, and skeletal system.
-
Thai Symphalangism Syndrome
Omim
Kantaputra et al. (2003) described a 12-year-old Thai girl with what they proposed represents a 'new' syndrome of proximal and distal symphalangism, postaxial polydactyly, hypodontia, and multiple and hyperplastic frenula. ... Mutation analyses of NOG (602991) and GDF5 (601146), the genes responsible for symphalangism-related syndromes, were negative. INHERITANCE - Isolated cases GROWTH Height - Short stature HEAD & NECK Head - Dolichocephaly Ears - Hypoplastic lobules - Hypoplastic helices Eyes - Blepharoptosis Nose - Prominent, broad nasal bridge - Broad philtrum Mouth - Multiple hyperplastic frenula - High-arched palate Teeth - Hypodontia SKELETAL Hands - Postaxial polydactyly - Symphalangism, proximal and distal - Brachydactyly Feet - Postaxial polydactyly - Brachydactyly - Congenital absence of distal phalanges (toes 2-5) ▲ Close
-
8q12 Microduplication Syndrome
Orphanet
The newly described 8q12 microduplication syndrome is associated with unusual and characteristic multi-organ clinical features, which include hearing loss, congenital heart defects, intellectual disability, hypotonia in infancy, and Duane anomaly (see this term). ... The 8q12 region includes CHD7 and it is proposed that this gene, associated with CHARGE syndrome by haploinsufficiency, causes a different phenotype by gain-of-dosage.
-
Retinohepatoendocrinologic Syndrome
Omim
Froyshov Larsen et al. (1978) described a new syndrome in 6 females in 2 sibships with a high degree of consanguinity and a male in another family. The syndrome consisted of total colorblindness from progressive cone dystrophy, degenerative liver disease, and endocrine dysfunction (hypothyroidism, 'maturity-onset diabetes of the young', repeated abortions or infertility).
-
Palant Cleft Palate Syndrome
Omim
Chromosomes were apparently normal. This is a type of syndrome that could be due to a small chromosomal aberration or even a repeated intrauterine insult. It is a type of syndrome that is difficult to categorize here for later recovery.
-
Stevenson-Carey Syndrome
Omim
Clinical Features Stevenson and Carey (2007) described 2 sisters with a multiple congenital anomaly syndrome with mental retardation, Pierre-Robin sequence, and cerebellar hypoplasia. ... Stevenson and Carey (2007) noted phenotypic overlap with the Ritscher-Schinzel (220210) and Toriello-Carey (217980) syndromes but concluded that the distinct pattern of malformations represented a provisionally unique syndrome. Craft et al. (2010) reported 2 sibs, a girl and a boy, born of consanguineous Pakistani parents, with features reminiscent of 3C syndrome. Both had delayed psychomotor development and dysmorphic facial features, including downslanting palpebral fissures, prominent nasal bridge, micrognathia, and small head size. ... Craft et al. (2010) noted that there is phenotypic variability in 3C syndrome, but also suggested some overlap with the phenotype in the 2 sisters reported by Stevenson and Carey (2007).
-
Marfanoid Hypermobility Syndrome
Omim
Very marked joint hypermobility and excessive stretchability of the skin suggested Ehlers-Danlos syndrome (EDS; see 130000), but no other features of that condition were present. The patient reported by Goodman et al. (1965, 1969) as having both EDS and Marfan syndrome probably had this disorder, and other possible examples are the cases of Roederer (1951) and Coventry (1961). ... Cotton and Brandt (1976) suggested that cardiovascular disease might be a significant feature of the marfanoid hypermobility syndrome. Handa et al. (2001) reported a 50-year-old Indian man with the unusual association of classic cutaneous features of Ehlers-Danlos syndrome, a marfanoid habitus, bladder diverticula, and multiple emphysematous bullae. ... The findings in this patient suggested that lung and bladder involvement may also be a part of this syndrome. Limbs - Arachnodactyly Inheritance - ?
-
Acromicric Dysplasia
Orphanet
Long-term follow-up shows that facial dysmorphism becomes less obvious in adults and that carpal tunnel syndrome is frequent in older patients. The entity known as Moore-Federman syndrome (see this term) characterized by short stature (with disproportionately short legs), joint stiffness, ocular abnormalities (hypermetropia, glaucoma) and thickened skin on the forearms, is believed to represent a variable clinical expression of acromicric dysplasia. ... Differential diagnosis Overlapping syndromes include geleophysic dysplasia, Weill-Marchesani syndrome, and Myhre syndrome (see these terms). Geleophysic dysplasia can be distinguished from acromicric dysplasia by the presence of cardiac abnormalities (e.g. cardiac valvular thickening), Weill-Marchesani syndrome by the presence of microspherophakia, and Myhre syndrome by the presence of prognathism, deafness, developmental delay, and a thick calvarium. ... Management and treatment Orthopedic management may be needed for hip dysplasia during childhood and for carpal tunnel syndrome in older patients. Physical therapy is required to prevent progressive joint limitation.
-
Hydrops-Ectopic Calcification-Moth-Eaten Skeletal Dysplasia
Wikipedia
"Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes" . Hum. Mol. Genet . 12. Spec No 1 (90001): R75–88. doi : 10.1093/hmg/ddg072 . ... External links [ edit ] Classification D ICD - 10 : Q77.3 OMIM : 215140 MeSH : C537299 External resources Orphanet : 1426 v t e Inborn errors of steroid metabolism Mevalonate pathway HMG-CoA lyase deficiency Hyper-IgD syndrome Mevalonate kinase deficiency To cholesterol 7-Dehydrocholesterol path: Hydrops-ectopic calcification-moth-eaten skeletal dysplasia CHILD syndrome Conradi-Hünermann syndrome Lathosterolosis Smith–Lemli–Opitz syndrome desmosterol path: Desmosterolosis Steroids Corticosteroid (including CAH ) aldosterone : Glucocorticoid remediable aldosteronism cortisol / cortisone : CAH 17α-hydroxylase CAH 11β-hydroxylase both: CAH 3β-dehydrogenase CAH 21-hydroxylase Apparent mineralocorticoid excess syndrome/11β-dehydrogenase Sex steroid To androgens 17α-Hydroxylase deficiency 17,20-Lyase deficiency Cytochrome b 5 deficiency 3β-Hydroxysteroid dehydrogenase deficiency 17β-Hydroxysteroid dehydrogenase deficiency 5α-Reductase deficiency Pseudovaginal perineoscrotal hypospadias To estrogens Aromatase deficiency Aromatase excess syndrome Other X-linked ichthyosis Antley–Bixler syndrome This article about an endocrine, nutritional, or metabolic disease is a stub .

-
Kahrizi Syndrome
Omim
A number sign (#) is used with this entry because Kahrizi syndrome (KHRZ) can be caused by homozygous mutation in the SRD5A3 gene (611715) on chromosome 4q12. Description Kahrizi syndrome is an autosomal recessive neurodevelopmental disorder characterized by mental retardation, cataracts, coloboma, kyphosis, and coarse facial features (summary by Kahrizi et al., 2009). ... Clinical Features Kahrizi et al. (2009) reported 3 Iranian sibs with a syndrome characterized by severe mental retardation, cataracts with onset in late adolescence, kyphosis, contractures of large joints, bulbous nose with broad nasal bridge, and thick lips. ... Mapping By linkage analysis, followed by haplotype analysis, of an Iranian family with syndromic mental retardation, Kahrizi et al. (2009) identified a 10.4-Mb region of homozygosity on chromosome 4p12-q12 between SNPs rs728293 and rs1105434 (parametric lod score of 3.38; nonparametric lod score of 4.53). Molecular Genetics In 3 sibs, born of consanguineous Iranian parents, with Kahrizi syndrome (Kahrizi et al., 2009), Kahrizi et al. (2011}) identified a homozygous truncating mutation in the SRD5A3 gene (611715.0006).
-
Multifocal Stenosing Ulceration Of The Small Intestine
Wikipedia
Differential diagnosis [ edit ] Idiopathic ulcerative jejunoileitis Lymphoma Infectious agents ( Campylobacter , Salmonella , Shigella , Yersinia and others) Infections in the immunosuppressed with unusual organisms Tropheryma whipplei , Mycobacterium avium intracellulare Drug induced (nonsteroidal anti-inflammatory drugs, gold , potassium , chemotherapy ) Zollinger-Ellison syndrome Heterotopic functioning gastric mucosa Meckel's diverticulum Crohn's disease Traumatic injury (surgical, seat belt injury, endoscopic biopsy, cautery during endoscopy, foreign body ingestion particularly batteries ) Ischemia Thrombotic diseases Degos disease Pseudoxanthoma elasticum Myeloproliferative disorders Antithrombin III deficiency Vasculitis Coeliac disease Behcet's disease Treatment [ edit ] Steroids seem to relieve the symptoms but long term treatment may be required. [2] Other immunosuppressants appear to be less effective. ... Arch Mal Appar Dig Mal Nutr. 1964;53:193–206 v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum

-
Trichorrhexis Nodosa
Wikipedia
Trichorrhexis nodosa Other names Hair shaft fracture [1] Tricho-hepato-enteric syndrome : Microscopic analysis of the hair shaft showing breaks located at nodes in the hair (trichorrhexis nodosa) and longitudinal breaks. ... In some cases, trichorrhexis nodosa may be caused by an underlying disorder such as argininosuccinic aciduria , Menkes' kinky hair syndrome , Netherton's syndrome , hypothyroidism , or trichothiodystrophy . ... References [ edit ] MedlinePlus Encyclopedia : Trichorrhexis nodosa External links [ edit ] Classification D ICD - 10 : L67.0 ICD - 9-CM : 704.2 DiseasesDB : 29680 External resources MedlinePlus : 001449 v t e Disorders of skin appendages Nail thickness: Onychogryphosis Onychauxis color: Beau's lines Yellow nail syndrome Leukonychia Azure lunula shape: Koilonychia Nail clubbing behavior: Onychotillomania Onychophagia other: Ingrown nail Anonychia ungrouped: Paronychia Acute Chronic Chevron nail Congenital onychodysplasia of the index fingers Green nails Half and half nails Hangnail Hapalonychia Hook nail Ingrown nail Lichen planus of the nails Longitudinal erythronychia Malalignment of the nail plate Median nail dystrophy Mees' lines Melanonychia Muehrcke's lines Nail–patella syndrome Onychoatrophy Onycholysis Onychomadesis Onychomatricoma Onychomycosis Onychophosis Onychoptosis defluvium Onychorrhexis Onychoschizia Platonychia Pincer nails Plummer's nail Psoriatic nails Pterygium inversum unguis Pterygium unguis Purpura of the nail bed Racquet nail Red lunulae Shell nail syndrome Splinter hemorrhage Spotted lunulae Staining of the nail plate Stippled nails Subungual hematoma Terry's nails Twenty-nail dystrophy Hair Hair loss / Baldness noncicatricial alopecia : Alopecia areata totalis universalis Ophiasis Androgenic alopecia (male-pattern baldness) Hypotrichosis Telogen effluvium Traction alopecia Lichen planopilaris Trichorrhexis nodosa Alopecia neoplastica Anagen effluvium Alopecia mucinosa cicatricial alopecia : Pseudopelade of Brocq Central centrifugal cicatricial alopecia Pressure alopecia Traumatic alopecia Tumor alopecia Hot comb alopecia Perifolliculitis capitis abscedens et suffodiens Graham-Little syndrome Folliculitis decalvans ungrouped: Triangular alopecia Frontal fibrosing alopecia Marie Unna hereditary hypotrichosis Hypertrichosis Hirsutism Acquired localised generalised patterned Congenital generalised localised X-linked Prepubertal Acneiform eruption Acne Acne vulgaris Acne conglobata Acne miliaris necrotica Tropical acne Infantile acne / Neonatal acne Excoriated acne Acne fulminans Acne medicamentosa (e.g., steroid acne ) Halogen acne Iododerma Bromoderma Chloracne Oil acne Tar acne Acne cosmetica Occupational acne Acne aestivalis Acne keloidalis nuchae Acne mechanica Acne with facial edema Pomade acne Acne necrotica Blackhead Lupus miliaris disseminatus faciei Rosacea Perioral dermatitis Granulomatous perioral dermatitis Phymatous rosacea Rhinophyma Blepharophyma Gnathophyma Metophyma Otophyma Papulopustular rosacea Lupoid rosacea Erythrotelangiectatic rosacea Glandular rosacea Gram-negative rosacea Steroid rosacea Ocular rosacea Persistent edema of rosacea Rosacea conglobata variants Periorificial dermatitis Pyoderma faciale Ungrouped Granulomatous facial dermatitis Idiopathic facial aseptic granuloma Periorbital dermatitis SAPHO syndrome Follicular cysts " Sebaceous cyst " Epidermoid cyst Trichilemmal cyst Steatocystoma simplex multiplex Milia Inflammation Folliculitis Folliculitis nares perforans Tufted folliculitis Pseudofolliculitis barbae Hidradenitis Hidradenitis suppurativa Recurrent palmoplantar hidradenitis Neutrophilic eccrine hidradenitis Ungrouped Acrokeratosis paraneoplastica of Bazex Acroosteolysis Bubble hair deformity Disseminate and recurrent infundibulofolliculitis Erosive pustular dermatitis of the scalp Erythromelanosis follicularis faciei et colli Hair casts Hair follicle nevus Intermittent hair–follicle dystrophy Keratosis pilaris atropicans Kinking hair Koenen's tumor Lichen planopilaris Lichen spinulosus Loose anagen syndrome Menkes kinky hair syndrome Monilethrix Parakeratosis pustulosa Pili ( Pili annulati Pili bifurcati Pili multigemini Pili pseudoannulati Pili torti ) Pityriasis amiantacea Plica neuropathica Poliosis Rubinstein–Taybi syndrome Setleis syndrome Traumatic anserine folliculosis Trichomegaly Trichomycosis axillaris Trichorrhexis ( Trichorrhexis invaginata Trichorrhexis nodosa ) Trichostasis spinulosa Uncombable hair syndrome Wooly hair nevus Sweat glands Eccrine Miliaria Colloid milium Miliaria crystalline Miliaria profunda Miliaria pustulosa Miliaria rubra Occlusion miliaria Postmiliarial hypohidrosis Granulosis rubra nasi Ross’ syndrome Anhidrosis Hyperhidrosis Generalized Gustatory Palmoplantar Apocrine Body odor Chromhidrosis Fox–Fordyce disease Sebaceous Sebaceous hyperplasia
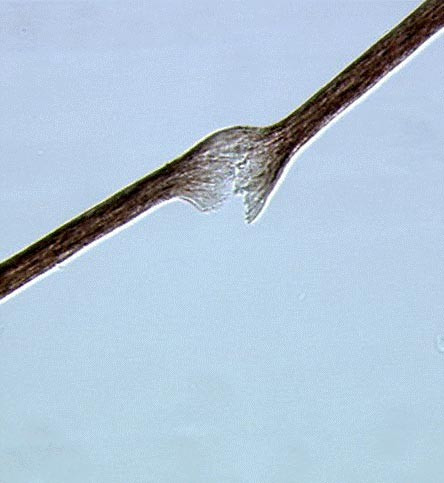
-
Urticarial Vasculitis
Wikipedia
WikiProject Molecular and Cellular Biology may be able to help recruit an expert. ( October 2011 ) Urticarial vasculitis Specialty Dermatology , immunology Urticarial vasculitis (also known as " chronic urticaria as a manifestation of venulitis ", " hypocomplementemic urticarial vasculitis syndrome ", " hypocomplementemic vasculitis " and " unusual lupus-like syndrome ") [1] is a skin condition characterized by fixed urticarial lesions that appear histologically as a vasculitis . [2] : 834 Contents 1 Mechanism 1.1 C1q 1.2 Impaired classical complement pathway 2 Diagnosis 3 Treatment 4 Popular culture 5 See also 6 References 7 External links Mechanism [ edit ] Antibodies are usually raised against foreign proteins, such as those made by a replicating virus or invading bacterium. ... There is no defined paradigm for the syndrome aetiology and severity in progression. ... External links [ edit ] Classification D ICD - 10 : L95.8 ( ILDS L95.810) External resources eMedicine : article/1085087 v t e Cutaneous vasculitis and other vascular-related cutaneous conditions Cutaneous vasculitis Erythema elevatum diutinum Capillaritis Urticarial vasculitis Nodular vasculitis Microvascular occlusion Calciphylaxis Cryoglobulinemic purpura / Cryoglobulinemic vasculitis vascular coagulopathy : Livedoid vasculitis Livedoid dermatitis Perinatal gangrene of the buttock Malignant atrophic papulosis Sneddon's syndrome Purpura Nonthrombocytopenic purpura : Cryofibrinogenemic purpura Drug-induced purpura Food-induced purpura IgA vasculitis Obstructive purpura Orthostatic purpura Purpura fulminans Purpura secondary to clotting disorders Purpuric agave dermatitis Pigmentary purpuric eruptions Solar purpura Traumatic purpura Waldenström hyperglobulinemic purpura Painful bruising syndrome ungrouped: Paroxysmal hand hematoma Postcardiotomy syndrome Deep vein thrombosis Superficial thrombophlebitis Mondor's disease Blueberry muffin baby Fibrinolysis syndrome Systemic vasculitis see Template:Systemic vasculitis Vascular malformations Arteriovenous malformation Bonnet–Dechaume–Blanc syndrome Cobb syndrome Parkes Weber syndrome Sinusoidal hemangioma lymphatic malformation Hennekam syndrome Aagenaes syndrome telangiectasia : Generalized essential telangiectasia Hereditary hemorrhagic telangiectasia Unilateral nevoid telangiectasia Ulcer Venous ulcer Arterial insufficiency ulcer Hematopoietic ulcer Neuropathic ulcer Acroangiodermatitis Lymphedema see Template:Lymphatic vessel disease Ungrouped vascular-related cutaneous conditions Raynaud's phenomenon Thromboangiitis obliterans Erythromelalgia Septic thrombophlebitis Arteriosclerosis obliterans Bier spots / Marshall–White syndrome Cholesterol embolus Reactive angioendotheliomatosis Trousseau's syndrome
-
Glucocorticoid Remediable Aldosteronism
Wikipedia
However, in subjects with glucocorticoid-remediable aldosteronism, ACTH increases the activity of existing aldosterone synthase , resulting in an abnormally high rate of aldosterone synthesis and hyperaldosteronism . [ citation needed ] Diagnosis [ edit ] Genetic testing [ citation needed ] Treatment [ edit ] In GRA, the hypersecretion of aldosterone and the accompanying hypertension are remedied when ACTH secretion is suppressed by administering glucocorticoids . [ citation needed ] Dexamethasone , spironolactone and eplerenone have been used in treatment. [4] See also [ edit ] Inborn errors of steroid metabolism Hyperaldosteronism Pseudohyperaldosteronism Apparent mineralocorticoid excess syndrome Aldosterone and aldosterone synthase References [ edit ] ^ Vonend O, Altenhenne C, Büchner NJ, et al. ... External links [ edit ] Classification D ICD - 10-CM : E26.02 ICD - 9-CM : 255.11 OMIM : 103900 MeSH : C563177 C563177, C563177 v t e Adrenal gland disorder Hyperfunction Aldosterone Hyperaldosteronism Primary aldosteronism Conn syndrome Bartter syndrome Glucocorticoid remediable aldosteronism AME Liddle's syndrome 17α CAH Pseudohypoaldosteronism Cortisol Cushing's syndrome Pseudo-Cushing's syndrome Steroid-induced osteoporosis Sex hormones 21α CAH 11β CAH Hypofunction Aldosterone Hypoaldosteronism 21α CAH 11β CAH Cortisol CAH Lipoid 3β 11β 17α 21α Sex hormones 17α CAH Inborn errors of steroid metabolism Adrenal insufficiency Adrenal crisis Adrenalitis Xanthogranulomatous Addison's disease Waterhouse–Friderichsen syndrome v t e Inborn errors of steroid metabolism Mevalonate pathway HMG-CoA lyase deficiency Hyper-IgD syndrome Mevalonate kinase deficiency To cholesterol 7-Dehydrocholesterol path: Hydrops-ectopic calcification-moth-eaten skeletal dysplasia CHILD syndrome Conradi-Hünermann syndrome Lathosterolosis Smith–Lemli–Opitz syndrome desmosterol path: Desmosterolosis Steroids Corticosteroid (including CAH ) aldosterone : Glucocorticoid remediable aldosteronism cortisol / cortisone : CAH 17α-hydroxylase CAH 11β-hydroxylase both: CAH 3β-dehydrogenase CAH 21-hydroxylase Apparent mineralocorticoid excess syndrome/11β-dehydrogenase Sex steroid To androgens 17α-Hydroxylase deficiency 17,20-Lyase deficiency Cytochrome b 5 deficiency 3β-Hydroxysteroid dehydrogenase deficiency 17β-Hydroxysteroid dehydrogenase deficiency 5α-Reductase deficiency Pseudovaginal perineoscrotal hypospadias To estrogens Aromatase deficiency Aromatase excess syndrome Other X-linked ichthyosis Antley–Bixler syndrome
-
Pityriasis Alba
Wikipedia
External links [ edit ] Classification D ICD - 10 : L30.5 ( ILDS L30.590) ICD - 9-CM : 696.5 DiseasesDB : 31121 External resources MedlinePlus : 001463 eMedicine : ped/1813 derm/333 emerg/425 v t e Diseases of the skin and appendages by morphology Growths Epidermal Wart Callus Seborrheic keratosis Acrochordon Molluscum contagiosum Actinic keratosis Squamous-cell carcinoma Basal-cell carcinoma Merkel-cell carcinoma Nevus sebaceous Trichoepithelioma Pigmented Freckles Lentigo Melasma Nevus Melanoma Dermal and subcutaneous Epidermal inclusion cyst Hemangioma Dermatofibroma (benign fibrous histiocytoma) Keloid Lipoma Neurofibroma Xanthoma Kaposi's sarcoma Infantile digital fibromatosis Granular cell tumor Leiomyoma Lymphangioma circumscriptum Myxoid cyst Rashes With epidermal involvement Eczematous Contact dermatitis Atopic dermatitis Seborrheic dermatitis Stasis dermatitis Lichen simplex chronicus Darier's disease Glucagonoma syndrome Langerhans cell histiocytosis Lichen sclerosus Pemphigus foliaceus Wiskott–Aldrich syndrome Zinc deficiency Scaling Psoriasis Tinea ( Corporis Cruris Pedis Manuum Faciei ) Pityriasis rosea Secondary syphilis Mycosis fungoides Systemic lupus erythematosus Pityriasis rubra pilaris Parapsoriasis Ichthyosis Blistering Herpes simplex Herpes zoster Varicella Bullous impetigo Acute contact dermatitis Pemphigus vulgaris Bullous pemphigoid Dermatitis herpetiformis Porphyria cutanea tarda Epidermolysis bullosa simplex Papular Scabies Insect bite reactions Lichen planus Miliaria Keratosis pilaris Lichen spinulosus Transient acantholytic dermatosis Lichen nitidus Pityriasis lichenoides et varioliformis acuta Pustular Acne vulgaris Acne rosacea Folliculitis Impetigo Candidiasis Gonococcemia Dermatophyte Coccidioidomycosis Subcorneal pustular dermatosis Hypopigmented Tinea versicolor Vitiligo Pityriasis alba Postinflammatory hyperpigmentation Tuberous sclerosis Idiopathic guttate hypomelanosis Leprosy Hypopigmented mycosis fungoides Without epidermal involvement Red Blanchable Erythema Generalized Drug eruptions Viral exanthems Toxic erythema Systemic lupus erythematosus Localized Cellulitis Abscess Boil Erythema nodosum Carcinoid syndrome Fixed drug eruption Specialized Urticaria Erythema ( Multiforme Migrans Gyratum repens Annulare centrifugum Ab igne ) Nonblanchable Purpura Macular Thrombocytopenic purpura Actinic/solar purpura Papular Disseminated intravascular coagulation Vasculitis Indurated Scleroderma / morphea Granuloma annulare Lichen sclerosis et atrophicus Necrobiosis lipoidica Miscellaneous disorders Ulcers Hair Telogen effluvium Androgenic alopecia Alopecia areata Systemic lupus erythematosus Tinea capitis Loose anagen syndrome Lichen planopilaris Folliculitis decalvans Acne keloidalis nuchae Nail Onychomycosis Psoriasis Paronychia Ingrown nail Mucous membrane Aphthous stomatitis Oral candidiasis Lichen planus Leukoplakia Pemphigus vulgaris Mucous membrane pemphigoid Cicatricial pemphigoid Herpesvirus Coxsackievirus Syphilis Systemic histoplasmosis Squamous-cell carcinoma v t e Dermatitis and eczema Atopic dermatitis Besnier's prurigo Seborrheic dermatitis Pityriasis simplex capillitii Cradle cap Contact dermatitis ( allergic , irritant ) plants: Urushiol-induced contact dermatitis African blackwood dermatitis Tulip fingers other: Abietic acid dermatitis Diaper rash Airbag dermatitis Baboon syndrome Contact stomatitis Protein contact dermatitis Eczema Autoimmune estrogen dermatitis Autoimmune progesterone dermatitis Breast eczema Ear eczema Eyelid dermatitis Topical steroid addiction Hand eczema Chronic vesiculobullous hand eczema Hyperkeratotic hand dermatitis Autosensitization dermatitis / Id reaction Candidid Dermatophytid Molluscum dermatitis Circumostomy eczema Dyshidrosis Juvenile plantar dermatosis Nummular eczema Nutritional deficiency eczema Sulzberger–Garbe syndrome Xerotic eczema Pruritus / Itch / Prurigo Lichen simplex chronicus / Prurigo nodularis by location: Pruritus ani Pruritus scroti Pruritus vulvae Scalp pruritus Drug-induced pruritus Hydroxyethyl starch-induced pruritus Senile pruritus Aquagenic pruritus Aquadynia Adult blaschkitis due to liver disease Biliary pruritus Cholestatic pruritus Prion pruritus Prurigo pigmentosa Prurigo simplex Puncta pruritica Uremic pruritus Other substances taken internally: Bromoderma Fixed drug reaction Nummular dermatitis Pityriasis alba Papuloerythroderma of Ofuji v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation
-
Abulia
Wikipedia
Differentiation from other disorders [ edit ] Both neurologists and psychiatrists recognize abulia to be a distinct clinical entity, but its status as a syndrome is unclear. Although abulia has been known to clinicians since 1838, it has been subjected to different interpretations – from 'a pure lack of will', in the absence of motor paralysis to, more recently, being considered 'a reduction in action emotion and cognition'. [6] As a result of the changing definition of abulia, there is currently a debate on whether or not abulia is a sign or a symptom of another disease, or its own disease that seems to appear in the presence of other more well-researched diseases, such as Alzheimer's disease. [6] A 2002 survey of two movement disorder experts, two neuropsychiatrists, and two rehabilitation experts, did not seem to shed any light on the matter of differentiating abulia from other DDMs. The experts used the terms " apathy " and "abulia" interchangeably and debated whether or not abulia was a discrete entity, or just a hazy gray area on a spectrum of more defined disorders. [6] Four of the experts said abulia was a sign and a symptom, and the group was split on whether or not it was a syndrome. [6] Another survey, which consisted of true and false questions about what abulia is distinct from, whether it is a sign, symptom, or syndrome, where lesions are present in cases of abulia, what diseases are commonly associated with abulia, and what current treatments are used for abulia, was sent to 15 neurologists and 10 psychiatrists . ... Yet again, there was disagreement about whether or not abulia is a sign, symptom, or syndrome. [6] [ citation needed ] The study of motivation has been mostly about how stimuli come to acquire significance for animals. ... This result supports the idea that abulia may exist independently of depression as its own syndrome. [11] Damage to anterior cingulate circuit [ edit ] The anterior cingulate circuit consists of the anterior cingulate cortex , also referred to as Brodmann area 24 , and its projections to the ventral striatum which includes the ventromedial caudate . ... External links [ edit ] The dictionary definition of abulia at Wiktionary v t e Symptoms , signs and syndromes associated with lesions of the brain and brainstem Brainstem Medulla (CN 8, 9, 10, 12) Lateral medullary syndrome/Wallenberg PICA Medial medullary syndrome/Dejerine ASA Pons (CN 5, 6, 7, 8) Upper dorsal pontine syndrome/Raymond-Céstan syndrome Lateral pontine syndrome ( AICA ) (lateral) Medial pontine syndrome / Millard–Gubler syndrome / Foville's syndrome ( basilar ) Locked-in syndrome Internuclear ophthalmoplegia One and a half syndrome Midbrain (CN 3, 4) Weber's syndrome ventral peduncle, PCA Benedikt syndrome ventral tegmentum, PCA Parinaud's syndrome dorsal, tumor Claude's syndrome Other Alternating hemiplegia Cerebellum Latearl Dysmetria Dysdiadochokinesia Intention tremor ) Medial Cerebellar ataxia Basal ganglia Chorea Dystonia Parkinson's disease Cortex ACA syndrome MCA syndrome PCA syndrome Frontal lobe Expressive aphasia Abulia Parietal lobe Receptive aphasia Hemispatial neglect Gerstmann syndrome Astereognosis Occipital lobe Bálint's syndrome Cortical blindness Pure alexia Temporal lobe Cortical deafness Prosopagnosia Thalamus Thalamic syndrome Other Upper motor neuron lesion Aphasia
-
Savant Syndrome
Wikipedia
"The savant syndrome: an extraordinary condition. A synopsis: past, present, future" . ... PMID 19528017 . ^ a b Miller LK (January 1999). "The savant syndrome: intellectual impairment and exceptional skill". ... PMID 9990844 . ^ Hughes JR (2012). "The savant syndrome and its possible relationship to epilepsy". ... Wisconsin Medical Society . ^ "Savant Syndrome Statistics" . Health Research Funding . 2014-07-12. ^ a b Conway, Richard; Carney, Daniel P. ... PMID 19528020 . ^ Mottron L, Dawson M, Soulières I (May 2009). "Enhanced perception in savant syndrome: patterns, structure and creativity" .




