-
Chromosome 5q Deletion Syndrome
Wikipedia
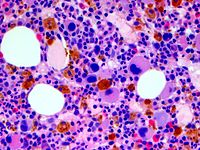
Human disease Chromosome 5q deletion syndrome Other names Chromosome 5q monosomy , 5q- syndrome Photomicrograph of bone marrow showing abnormal mononuclear megakaryocytes typical of 5q- syndrome Specialty Hematology Chromosome 5q deletion syndrome is an acquired, hematological disorder characterized by loss of part of the long arm ( q arm , band 5q33.1) of human chromosome 5 in bone marrow myelocyte cells. ... "Identification of RPS14 as a 5q- syndrome gene by RNA interference screen" (PDF) . ... S2CID 7987486 . ^ Online Mendelian Inheritance in Man (OMIM): 5q- syndrome - 153550 ^ List A; Dewald G; Bennett J; et al. ... "Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion". N. ... S2CID 38251104 . External links [ edit ] 5q- syndrome at NIH 's Office of Rare Diseases Classification D ICD-O : M9986/3 OMIM : 153550 MeSH : C535323 DiseasesDB : 34573 v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22 v t e Myeloid -related hematological malignancy CFU-GM / and other granulocytes CFU-GM Myelocyte AML : Acute myeloblastic leukemia M0 M1 M2 APL/M3 MP Chronic neutrophilic leukemia Monocyte AML AMoL/M5 Myeloid dendritic cell leukemia CML Philadelphia chromosome Accelerated phase chronic myelogenous leukemia Myelomonocyte AML M4 MD-MP Juvenile myelomonocytic leukemia Chronic myelomonocytic leukemia Other Histiocytosis CFU-Baso AML Acute basophilic CFU-Eos AML Acute eosinophilic MP Chronic eosinophilic leukemia / Hypereosinophilic syndrome MEP CFU-Meg MP Essential thrombocytosis Acute megakaryoblastic leukemia CFU-E AML Erythroleukemia/M6 MP Polycythemia vera MD Refractory anemia Refractory anemia with excess of blasts Chromosome 5q deletion syndrome Sideroblastic anemia Paroxysmal nocturnal hemoglobinuria Refractory cytopenia with multilineage dysplasia CFU-Mast Mastocytoma Mast cell leukemia Mast cell sarcoma Systemic mastocytosis Mastocytosis : Diffuse cutaneous mastocytosis Erythrodermic mastocytosis Adult type of generalized eruption of cutaneous mastocytosis Urticaria pigmentosa Mast cell sarcoma Solitary mastocytoma Systemic mastocytosis Xanthelasmoidal mastocytosis Multiple/unknown AML Acute panmyelosis with myelofibrosis Myeloid sarcoma MP Myelofibrosis Acute biphenotypic leukaemiaRPS14, TP53, MIR145, MIR146A, SPARC, TRPM1, RARS1, CSF1R, IL12B, GLRA1, FGF1, F2R, EGR1, CD74, TTN, KLF1, LARP1, THOP1, SH3TC2, ATXN1, S100A8, TRPM3, NPM1, MYD88, MSX2, ANXA5, KITLG, FLI1, CD14, CD34, CCR7, CSF2, CSNK1A1, EPO, MECOM, HMMR, JAK2, HSPA4, HSPA9, HTC2, IL3, IL5, ATOX1, IRF1, LINC00273
-
Chromosome Instability Syndrome
Wikipedia
Chromosome instability syndromes are a group of inherited conditions associated with chromosomal instability and breakage. They often lead to an increased tendency to develop certain types of malignancies. [1] The following chromosome instability syndromes are known: Ataxia telangiectasia Ataxia telangiectasia-like disorder Bloom syndrome Fanconi anaemia Nijmegen breakage syndrome Neurodegenerative diseases [ edit ] Chromosome instability syndromes include several inherited neurodegenerative diseases that are due to mutations in genes that encode enzymes necessary for DNA repair . Epigenetic alterations often occur in association with the DNA repair defect, and such alterations likely have a role in the etiology of the disease. Chromosome instability syndromes due to impaired DNA repair and with features of neurodegeneration and epigenetic alteration were summarized by Bernstein and Bernstein. [ citation needed ] These syndromes include Aicardi-Goutieres syndrome , amyotrophic lateral sclerosis , ataxia-telangiectasia , Cockayne syndrome , fragile X syndrome , Friedrich's ataxia , Huntington's disease , spinocerebellar ataxia type 1 , trichothiodystrophy and xeroderma pigmentosum . ... "Chromosome instability syndromes" . Best Pract Res Clin Haematol . 14 (3): 631–44. doi : 10.1053/beha.2001.0158 . ... PMID 27802094 . v t e Metabolic disease : DNA replication and DNA repair-deficiency disorder DNA replication Separation/initiation: RNASEH2A Aicardi–Goutières syndrome 4 Termination/ telomerase : DKC1 Dyskeratosis congenita DNA repair Nucleotide excision repair Cockayne syndrome / DeSanctis–Cacchione syndrome Thymine dimer Xeroderma pigmentosum IBIDS syndrome MSI / DNA mismatch repair Hereditary nonpolyposis colorectal cancer Muir–Torre syndrome Mismatch repair cancer syndrome MRN complex Ataxia telangiectasia Nijmegen breakage syndrome Other RecQ helicase Bloom syndrome Werner syndrome Rothmund–Thomson syndrome / Rapadilino syndrome Fanconi anemia Li-Fraumeni syndrome Severe combined immunodeficiency
-
Kindler Syndrome
Wikipedia
Kindler syndrome Other names Congenital poikiloderma with blisters and keratoses, [1] Congenital poikiloderma with bullae and progressive cutaneous atrophy, [1] Hereditary acrokeratotic poikiloderma, [1] Hyperkeratosis–hyperpigmentation syndrome, [2] : 511 Acrokeratotic poikiloderma, Weary–Kindler syndrome [3] : 558 Kindler syndrome has an autosomal recessive pattern of inheritance. ... As individuals with Kindler syndrome age, they tend to have fewer problems with blistering and photosensitivity. However, pigment changes and thinning of the skin become more prominent. [4] Kindler syndrome can affect various mucous tissues such as the mouth and eyes, which can lead to other health problems. [5] Genetics [ edit ] Kindler syndrome is an autosomal recessive genodermatosis . ... S2CID 221861310 . ^ a b "Kindler syndrome" . Genetics Home Reference . NIH . ... External links [ edit ] Classification D ICD - 10 : Q82.8 OMIM : 173650 MeSH : C536321 DiseasesDB : 32778 External resources eMedicine : derm/943 Orphanet : 2908 v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark v t e Cell membrane protein disorders (other than Cell surface receptor , enzymes , and cytoskeleton ) Arrestin Oguchi disease 1 Myelin Pelizaeus–Merzbacher disease Dejerine–Sottas disease Charcot–Marie–Tooth disease 1B, 2J Pulmonary surfactant Surfactant metabolism dysfunction 1, 2 Cell adhesion molecule IgSF CAM : OFC7 Cadherin : DSG1 Striate palmoplantar keratoderma 1 DSG2 Arrhythmogenic right ventricular dysplasia 10 DSG4 LAH1 DSC2 Arrhythmogenic right ventricular dysplasia 11 Integrin : cell surface receptor deficiencies Tetraspanin TSPAN7 X-Linked mental retardation 58 TSPAN12 Familial exudative vitreoretinopathy 5 Other KIND1 Kindler syndrome HFE HFE hereditary haemochromatosis DYSF Distal muscular dystrophy Limb-girdle muscular dystrophy 2B See also other cell membrane proteinsFERMT1, ACTB, FBLIM1, FERMT2, UVRAG, COMMD3-BMI1, FERMT3, CKAP4, TP63, XPC, RPE65, BMI1, PXN, MAPK8, PLEK, KRT5, COL7A1, CDKN2A, CDK2, CDK1, H3P10

-
Hearing Loss With Craniofacial Syndromes
Wikipedia
Contents 1 Treacher Collins syndrome 2 Pierre Robin sequence 3 Stickler syndrome 4 Apert syndrome 5 Crouzon syndrome 6 Pfeiffer syndrome 7 Ectrodactyly–ectrodermal dysplasia–cleft syndrome 8 Saethre–Chotzen syndrome 9 Velocardiofacial syndrome 10 Hemifacial microsomia 11 Nager syndrome 12 See also 13 References 14 External links Treacher Collins syndrome [ edit ] Main article: Treacher Collins syndrome Individuals with Treacher Collins syndrome often have both cleft palate and hearing loss, in addition to other disabilities. ... This mildly progressive sensorineural loss, or more significant losses (associated with Types II and III Stickler syndrome) is present in about 80% of patients with Stickler syndrome. ... Otol Neurotol. 2009 Feb;30(2):184-9) Crouzon syndrome [ edit ] Main article: Crouzon syndrome Patients with Crouzon syndrome sometimes exhibit malformations of the external ear and/or the middle ear, such as malalignment of the pinna (Peterson-Falzone et al. , 2001). ... Ectrodactyly–ectrodermal dysplasia–cleft syndrome [ edit ] Main article: Ectrodactyly–ectodermal dysplasia–cleft syndrome Conductive hearing loss has been reported by many with ectrodactyly–ectodermal dysplasia–cleft (EEC) syndrome in association with a cleft palate (Perterson-Falzone, 2001). Saethre–Chotzen syndrome [ edit ] Main article: Saethre–Chotzen syndrome In Saethre–Chotzen syndrome, the ears may be low set, posteriorly rotated, have other minor anomalies and there may be a presence of a conductive hearing loss or a mixed hearing loss (Perterson-Falszone, 2001).
-
Alport Syndrome
Wikipedia
Alport syndrome Hearing loss effect of Alport syndrome in 13-year-old boy Specialty Medical genetics Alport syndrome is a genetic disorder [1] affecting around 1 in 5,000-10,000 children, [2] characterized by glomerulonephritis , end-stage kidney disease, and hearing loss. [3] Alport syndrome can also affect the eyes, though the changes do not usually affect sight, except when changes to the lens occur in later life. ... Retrieved 2004-06-16 . ^ "What is Alport syndrome?" . Alport syndrome . Retrieved 2019-01-06 . ^ " Alport syndrome " at Dorland's Medical Dictionary ^ Lagona E, Tsartsali L, Kostaridou S, Skiathitou A, Georgaki E, Sotsiou F (April 2008). "Skin biopsy for the diagnosis of Alport syndrome" . Hippokratia . 12 (2): 116–8. ... PMID 18343956 . S2CID 28650514 . ^ Alport Syndrome~treatment at eMedicine ^ "Alport syndrome" . ... This article incorporates public domain material from the United States National Library of Medicine document: "Alport syndrome" . (Genetics Home Reference) External links [ edit ] Classification D ICD - 10 : Q87.8 ICD - 9-CM : 759.89 OMIM : 301050 104200 203780 300195 MeSH : y D009394 y DiseasesDB : 454 External resources MedlinePlus : 000504 eMedicine : med/110 Patient UK : Alport syndrome GeneReviews : Collagen IV-Related Nephropathies (Alport Syndrome and Thin Basement Membrane Nephropathy) GeneReview/NIH/UW entry on Alport syndrome v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins v t e Congenital malformations and deformations of urinary system Abdominal Kidney Renal agenesis / Potter sequence , Papillorenal syndrome cystic Polycystic kidney disease Meckel syndrome Multicystic dysplastic kidney Medullary sponge kidney Horseshoe kidney Renal ectopia Nephronophthisis Supernumerary kidney Pelvic kidney Dent's disease Alport syndrome Ureter Ectopic ureter Megaureter Duplicated ureter Pelvic Bladder Bladder exstrophy Urethra Epispadias Hypospadias Posterior urethral valves Penoscrotal transposition Vestigial Urachus Urachal cyst Urachal fistula Urachal sinus v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia

-
Constitutional Mismatch Repair Deficiency Syndrome
Medlineplus
Almost all people with CMMRD syndrome develop cancer before age 18, generally in late childhood. ... A single mutation in any of the genes associated with CMMRD syndrome generally leads to a different cancer predisposition syndrome called Lynch syndrome. Because the parents of an individual with CMMRD syndrome typically have a single copy of the mutated gene, they may have Lynch syndrome. ... Unlike CMMRD syndrome, individuals with Lynch syndrome often develop these cancers in adulthood. Additionally, not all people with Lynch syndrome develop cancerous tumors, so a person with CMMRD syndrome might not have a history of cancer in their family.
-
Ring Chromosome 14 Syndrome
Wikipedia
Ring chromosome 14 syndrome Other names Ring 14, Ring chromosome 14 Formation of a ring chromosome. ... "ring chromosome 14 syndrome" . Genetics Home Reference . Retrieved 2017-03-17 . ^ a b c d e Disorders, National Organization for Rare (2003). ... "The ring 14 syndrome". European Journal of Medical Genetics . 55 (5): 374–380. doi : 10.1016/j.ejmg.2012.03.009 . ... Zollino M.; Seminara L.; Orteschi D.; Gobbi G.; Giovannini S.; Della Giustina E.; Frattini D.; Scarano A.; Neri G. (2009). "The Ring14 Syndrome: Clinical and Molecular Definition" (PDF) . ... External links [ edit ] Classification D ICD - 10 : Q93.2 OMIM : 616606 MeSH : C535487 DiseasesDB : 34753 External resources Orphanet : 1440 Scholia has a topic profile for Ring chromosome 14 syndrome . v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22

-
Microdeletion Syndrome
Wikipedia
Larger chromosomal deletion syndromes are detectable using karyotyping techniques. Examples [ edit ] DiGeorge syndrome or velocardiofacial syndrome [3] – most common microdeletion syndrome Prader–Willi syndrome [4] [5] Angelman syndrome [4] Neurofibromatosis type 1 [6] Neurofibromatosis type II [7] [8] Williams syndrome [9] Miller–Dieker syndrome [10] Smith–Magenis syndrome [11] Rubinstein–Taybi syndrome [12] Wolf–Hirschhorn syndrome [13] References [ edit ] ^ H. ... ISBN 0-7216-9347-4 . ^ "Microdeletion syndrome" . Genetics Home Reference. 17 April 2014 . ... (Sep 13, 2002). "Ch 19. Microdeletion Syndromes: Characteristics and Diagnosis". ... ISBN 978-0470016176 . 13 chromosomal disorders you may not have heard of v t e Mutation Mechanisms of mutation Insertion Deletion Substitution Transversion Transition Mutation with respect to structure Point mutation Nonsense mutation Missense mutation Conservative mutation Silent mutation Frameshift mutation Dynamic mutation Large-scale mutation Chromosomal translocations Chromosomal inversions Mutation with respect to overall fitness Deleterious mutation Advantageous mutation Neutral mutation Nearly neutral mutation Synonymous mutation Nonsynonymous mutation v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22
-
Overlap Syndrome
Wikipedia
Overlap syndrome Specialty Rheumatology An overlap syndrome is a medical condition which shares features of at least two more widely recognised disorders. ... An overlap syndrome can be seen whereby a mutation in the SCN5A gene encoding the cardiac sodium channel causes a reduction in the peak sodium current leading to the typical ECG features of Brugada syndrome, but which simultaneously increases the sustained late sodium current leading to the ECG features of Long QT syndrome type 3 . [5] Brugada syndrome can also overlap with arrhythmogenic cardiomyopathy due to certain mutations in the plakophilin gene. [6] See also [ edit ] Autoimmune Mixed connective tissue disease References [ edit ] ^ Maddison PJ (December 1991). "Overlap syndromes and mixed connective tissue disease". ... "Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy" . ... Further reading [ edit ] The Myositis Association Overlap Syndrome 2011 Conference Presentation External links [ edit ] Classification D ICD - 10 : M35.1 v t e Systemic connective tissue disorders General Systemic lupus erythematosus Drug-induced SLE Libman–Sacks endocarditis Inflammatory myopathy Myositis Dermatopolymyositis Dermatomyositis / Juvenile dermatomyositis Polymyositis * Inclusion body myositis Scleroderma Systemic scleroderma Progressive systemic sclerosis CREST syndrome Overlap syndrome / Mixed connective tissue disease Other hypersensitivity / autoimmune Sjögren syndrome Other Behçet's disease Polymyalgia rheumatica Eosinophilic fasciitis Eosinophilia–myalgia syndrome fibrillin Marfan syndrome Congenital contractural arachnodactylySCN5A, EXOSC10, COPD, SMAD4, TET2, MEFV, DLAT, CRH, ND1, ND5, TNF, TPO, PRPF8, TTN, TRIM21, SNRNP70, VWF, RUVBL1, MCF2L, C1QL1, PUF60, AIPL1, PTPN22, ADAMTSL4, RNPC3, ENAH, LSM2, NLRP3, RBM45, PLF, FBXW4, AADAC, RYR2, FLNA, AQP4, CALR, CBL, CDH1, CMTX3, COL1A1, COL12A1, CRP, DDOST, DNMT3A, GUCA1A, RPE65, HLA-DRB1, HRES1, IL6, CXCL8, JAK2, MPO, CYTB, PECAM1, ABL1, RAB4A, PSC
-
Worth Syndrome
Wikipedia
Worth syndrome Worth syndrome has an autosomal dominant pattern of inheritance. Worth syndrome , also known as benign form of Worth hyperostosis corticalis generalisata with torus platinus , autosomal dominant osteosclerosis , autosomal dominant endosteal hyperostosis or Worth disease , [1] [2] is a rare autosomal dominant congenital disorder that is caused by a mutation in the LRP5 gene. [3] It is characterized by increased bone density and benign bony structures on the palate . [1] [3] [4] [5] Contents 1 Causes 2 Diagnosis 3 Treatment 4 History 5 References 6 External links Causes [ edit ] Worth syndrome is caused by a mutation in the LRP5 gene, located on human chromosome 11q13.4 . [3] [6] The disorder is inherited in an autosomal dominant fashion. [1] This indicates that the defective gene responsible for a disorder is located on an autosome (chromosome 11 is an autosome), and only one copy of the defective gene is sufficient to cause the disorder, when inherited from a parent who has the disorder. ... Gorlin and L. Glass, distinguished the syndrome from van Buchem disease . In 1987 a group of Spanish doctors pointed out that the condition may not be benign , and may sometimes cause nerve damage. [1] References [ edit ] ^ a b c d Online Mendelian Inheritance in Man (OMIM): 144750 ^ Diseases Database (DDB): 32107 ^ a b c Van Wesenbeeck L, Cleiren E, Gram J, et al. ... PMC 1180253 . PMID 12579474 . ^ "Worth Syndrome" . Retrieved September 12, 2010 . ^ "Worth's Syndrome" . ... Retrieved September 12, 2010 . ^ Online Mendelian Inheritance in Man (OMIM): 603506 External links [ edit ] Classification D ICD - 10 : Q78.2 OMIM : 144750 DiseasesDB : 32107 External resources Orphanet : 2790 v t e Cell surface receptor deficiencies G protein-coupled receptor (including hormone ) Class A TSHR ( Congenital hypothyroidism 1 ) LHCGR ( Luteinizing hormone insensitivity , Leydig cell hypoplasia , Male-limited precocious puberty ) FSHR ( Follicle-stimulating hormone insensitivity , XX gonadal dysgenesis ) GnRHR ( Gonadotropin-releasing hormone insensitivity ) EDNRB ( ABCD syndrome , Waardenburg syndrome 4a , Hirschsprung's disease 2 ) AVPR2 ( Nephrogenic diabetes insipidus 1 ) PTGER2 ( Aspirin-induced asthma ) Class B PTH1R ( Jansen's metaphyseal chondrodysplasia ) Class C CASR ( Familial hypocalciuric hypercalcemia ) Class F FZD4 ( Familial exudative vitreoretinopathy 1 ) Enzyme-linked receptor (including growth factor ) RTK ROR2 ( Robinow syndrome ) FGFR1 ( Pfeiffer syndrome , KAL2 Kallmann syndrome ) FGFR2 ( Apert syndrome , Antley–Bixler syndrome , Pfeiffer syndrome , Crouzon syndrome , Jackson–Weiss syndrome ) FGFR3 ( Achondroplasia , Hypochondroplasia , Thanatophoric dysplasia , Muenke syndrome ) INSR ( Donohue syndrome Rabson–Mendenhall syndrome ) NTRK1 ( Congenital insensitivity to pain with anhidrosis ) KIT ( KIT Piebaldism , Gastrointestinal stromal tumor ) STPK AMHR2 ( Persistent Müllerian duct syndrome II ) TGF beta receptors : Endoglin / Alk-1 / SMAD4 ( Hereditary hemorrhagic telangiectasia ) TGFBR1 / TGFBR2 ( Loeys–Dietz syndrome ) GC GUCY2D ( Leber's congenital amaurosis 1 ) JAK-STAT Type I cytokine receptor : GH ( Laron syndrome ) CSF2RA ( Surfactant metabolism dysfunction 4 ) MPL ( Congenital amegakaryocytic thrombocytopenia ) TNF receptor TNFRSF1A ( TNF receptor associated periodic syndrome ) TNFRSF13B ( Selective immunoglobulin A deficiency 2 ) TNFRSF5 ( Hyper-IgM syndrome type 3 ) TNFRSF13C ( CVID4 ) TNFRSF13B ( CVID2 ) TNFRSF6 ( Autoimmune lymphoproliferative syndrome 1A ) Lipid receptor LRP : LRP2 ( Donnai–Barrow syndrome ) LRP4 ( Cenani–Lenz syndactylism ) LRP5 ( Worth syndrome , Familial exudative vitreoretinopathy 4 , Osteopetrosis 1 ) LDLR ( LDLR Familial hypercholesterolemia ) Other/ungrouped Immunoglobulin superfamily : AGM3, 6 Integrin : LAD1 Glanzmann's thrombasthenia Junctional epidermolysis bullosa with pyloric atresia EDAR ( EDAR hypohidrotic ectodermal dysplasia ) PTCH1 ( Nevoid basal-cell carcinoma syndrome ) BMPR1A ( BMPR1A juvenile polyposis syndrome ) IL2RG ( X-linked severe combined immunodeficiency ) See also cell surface receptors

-
Ellis–van Creveld Syndrome
Wikipedia
Ellis–van Creveld syndrome Other names Chondroectodermal dysplasia Polydactyly in Ellis–van Creveld syndrome Specialty Medical genetics Ellis–van Creveld syndrome (also called mesoectodermal dysplasia but see ' Nomenclature ' section below) is a rare genetic disorder of the skeletal dysplasia type. ... The cilia defects adversely affect "numerous critical developmental signaling pathways" essential to cellular development and thus offer a plausible hypothesis for the often multi-symptom nature of a large set of syndromes and diseases. Known ciliopathies include primary ciliary dyskinesia , Bardet–Biedl syndrome , polycystic kidney and liver disease , nephronophthisis , Alström syndrome , Meckel–Gruber syndrome and some forms of retinal degeneration . [5] Weyers acrofacial dysostosis is due to another mutation in the EVC gene and hence is allelic with Ellis–van Creveld syndrome. [6] Treatment [ edit ] There is no causative / curative therapy. ... Retrieved 7 April 2013 . ^ a b Ellis–van Creveld syndrome . Genes and Diseases . NCBI. 1998 . ... "Mutations in a new gene in Ellis–van Creveld syndrome and Weyers acrodental dysostosis". ... External links [ edit ] Classification D ICD - 10 : Q77.6 ICD - 9-CM : 756.55 OMIM : 225500 MeSH : D004613 DiseasesDB : 29309 External resources MedlinePlus : 001667 eMedicine : ped/660 Orphanet : 289 v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark

-
Keutel Syndrome
Wikipedia
Keutel syndrome Other names Pulmonic stenosis-brachytelephalangism-calcification of cartilages syndrome [1] Keutel syndrome has an autosomal recessive pattern of inheritance. ... "Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome". Nat. Genet . 21 (1): 142–4. doi : 10.1038/5102 . ... S2CID 6833062 . ^ Cormode EJ, Dawson M, Lowry RB (1986). "Keutel syndrome: clinical report and literature review". ... "Tracheobronchial stenosis in Keutel syndrome" . European Respiratory Journal . 17 (3): 566–569. doi : 10.1183/09031936.01.17305660 . ... S2CID 25194009 . External links [ edit ] Keutel syndrome at NIH 's Office of Rare Diseases Classification D ICD - 10 : Q87.8 OMIM : 245150 MeSH : C536167 DiseasesDB : 33698 External resources Orphanet : 85202 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome
-
49,xxxxy
Wikipedia
It can be considered a form or variant of Klinefelter syndrome (47,XXY). [7] Individuals with this syndrome are typically mosaic , being 49,XXXXY/48, XXXX. [4] It is genetic but not hereditary, meaning that while the genes of the parents cause the syndrome, there is a small chance of more than one child having the syndrome. ... As a result of infertility, one man from Iran used artificial reproductive methods. [4] An infant in Iran diagnosed with 49,XXXXY syndrome was born with patent ductus arteriosus , which was corrected with surgery, and other complications that were managed with replacement therapy. [4] See also [ edit ] Aneuploidy Turner syndrome Klinefelter syndrome 49, XXXXX , a similar syndrome that affects females References [ edit ] ^ Visootsak J, Graham JM (2006). "Klinefelter syndrome and other sex chromosomal aneuploidies" . ... "Neonatal diagnosis of 49, XXXXY syndrome" . Iranian Journal of Reproductive Medicine . 13 (3): 181–184. ... External links [ edit ] Classification D ICD - 9-CM : 758.81 DiseasesDB : 32552 49 XXXXY at the National Organization of Rare Diseases v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22
-
Mass Syndrome
Wikipedia
MASS syndrome Other names Mitral valve-aorta-skeleton-skin syndrome [1] This condition is inherited in an autosomal dominant manner Specialty Medical condition MASS syndrome is a medical disorder of the connective tissue similar to Marfan syndrome . ... Treatment options for MASS syndrome are largely determined on a case-by-case basis and generally address the symptoms as opposed to the actual disorder; [5] furthermore, due to the similarities between these two disorders, individuals with MASS syndrome follow the same treatment plans as those with Marfan syndrome. [6] Other possible symptoms are mitral valve prolapse , a large aortic root diameter, and myopia . [2] The skeletal features found in MASS syndrome include curvature of the spine ( scoliosis ), chest wall deformities, and joint hypermobility . [2] MASS syndrome and Marfan syndrome are overlapping connective tissue disorders . ... Skin involvement in MASS syndrome is typically limited to stretch marks ( striae distensae ). ... "Orphanet: MASS syndrome" . www.orpha.net . Retrieved 27 August 2019 . ^ a b c d "MASS PHENOTYPE" . ... "Evaluation of the adolescent or adult with some features of Marfan syndrome" . Genet. Med . 14 (1): 171–7. doi : 10.1038/gim.2011.48 .FBN1, LRP5, SOST, DKK1, AHR, TPO, WNT3, WNT11, MBTPS1, FERMT2, SIRT1, ABCB4, FGF21, ANKH, SMURF1, SMURF2, NEGR1, CRNDE, SLIT3, PGR, ANK1, LEP, HIF1A, FGFR1, F2R, EPAS1, S1PR3, CYP1B1, CTNNB1, CRK, COL1A1, CNR1, CALCR, ARSB, AR, SNORD116@
-
Sneddon's Syndrome
Wikipedia
Sneddon syndrome Other names Ehrmann-Sneddon syndrome, Livedo racemosa-cerebrovascular accident syndrome, Livedo reticularis-cerebrovascular accident syndrome [1] This condition is inherited in an autosomal recessive manner [2] Specialty Rheumatology Sneddon's syndrome [1] is a form of arteriopathy characterized by several symptoms, including: Severe, transient neurological symptoms or stroke Livedo reticularis , or livedo racemosa Contents 1 Signs and symptoms 2 Pathogenesis 3 Diagnosis 4 Treatment 5 Epidemiology 6 History 7 See also 8 References 9 Further reading 10 External links Signs and symptoms [ edit ] Sneddon's syndrome generally manifests with stroke or severe, transient neurological symptoms, and a skin rash ( livedo reticularis ). ... About 40-60% of patients with the syndrome test positive for antiphospholipid antibodies. ... Sneddon's Syndrome most often becomes apparent in women in their thirties, though cases do occur in men and in children. ... See also [ edit ] Livedoid vasculopathy List of cutaneous conditions References [ edit ] ^ a b c Berlit, Peter. "Sneddon's Syndrome" . Orphanet . ^ "OMIM Entry - # 182410 - SNEDDON SYNDROME" . omim.org . ... External links [ edit ] Classification D ICD - 10 : M30.8 ( ILDS M30.820) OMIM : 182410 MeSH : D018860 DiseasesDB : 12257 v t e Cerebrovascular diseases including stroke Ischaemic stroke Brain Anterior cerebral artery syndrome Middle cerebral artery syndrome Posterior cerebral artery syndrome Amaurosis fugax Moyamoya disease Dejerine–Roussy syndrome Watershed stroke Lacunar stroke Brain stem Brainstem stroke syndrome Medulla Medial medullary syndrome Lateral medullary syndrome Pons Medial pontine syndrome / Foville's Lateral pontine syndrome / Millard-Gubler Midbrain Weber's syndrome Benedikt syndrome Claude's syndrome Cerebellum Cerebellar stroke syndrome Extracranial arteries Carotid artery stenosis precerebral Anterior spinal artery syndrome Vertebrobasilar insufficiency Subclavian steal syndrome Classification Brain ischemia Cerebral infarction Classification Transient ischemic attack Total anterior circulation infarct Partial anterior circulation infarct Other CADASIL Binswanger's disease Transient global amnesia Haemorrhagic stroke Extra-axial Epidural Subdural Subarachnoid Cerebral/Intra-axial Intraventricular Brainstem Duret haemorrhages General Intracranial hemorrhage Aneurysm Intracranial aneurysm Charcot–Bouchard aneurysm Other Cerebral vasculitis Cerebral venous sinus thrombosis v t e Systemic vasculitis Large vessel Takayasu's arteritis Giant cell arteritis Medium vessel Polyarteritis nodosa Kawasaki disease Thromboangiitis obliterans Small vessel Pauci-immune c-ANCA Granulomatosis with polyangiitis p-ANCA Eosinophilic granulomatosis with polyangiitis Microscopic polyangiitis Type III hypersensitivity Cutaneous small-vessel vasculitis IgA vasculitis Ungrouped Acute hemorrhagic edema of infancy Cryoglobulinemic vasculitis Bullous small vessel vasculitis Cutaneous small-vessel vasculitis Other Goodpasture syndrome Sneddon's syndrome

-
Haber Syndrome
Wikipedia
Haber syndrome Specialty Dermatology Haber syndrome is a cutaneous disorder of hyperpigmentation characterized by reticulated pigmentation of the person's skin. [1] A rare genodermatosis, [2] its key features include "rosacea-like facial eruption[,] reticulated hyperpigmentation of major flexures, comedones on the back and neck, and pitted facial scars." [1] See also [ edit ] List of cutaneous conditions References [ edit ] ^ a b Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... ISBN 978-1-4160-2999-1 . ^ McCormack, CJ; Cowen P. (May 1997). "Haber's syndrome". Australas. J. Dermatol . 38 (2): 82–4. doi : 10.1111/j.1440-0960.1997.tb01113.x . ... External links [ edit ] Classification D DiseasesDB : 31372 v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation This cutaneous condition article is a stub .
-
Autosomal Recessive Cerebellar Ataxia
Wikipedia
It may refer to: Autosomal recessive cerebellar ataxia type 1 , a.k.a. autosomal recessive ataxia, Beauce type Autosomal recessive cerebelloparenchymal disorder type 3 Dysequilibrium syndrome CAMOS syndrome Cerebellar ataxia, Cayman type Joubert syndrome with oculorenal defect Joubert syndrome Joubert syndrome with hepatic defect Orofaciodigital syndrome type 6 Joubert syndrome with ocular defect Joubert syndrome with renal defect Joubert syndrome with Jeune asphyxiating thoracic dystrophy Autosomal recessive cerebellar ataxia due to CWF19L1 deficiency Congenital cerebellar ataxia due to RNU12 mutation Ataxia with vitamin E deficiency Abetalipoproteinemia Refsum disease Cerebrotendinous xanthomatosis Infantile Refsum disease Recessive mitochondrial ataxia syndrome Autosomal recessive ataxia due to PEX10 deficiency Autosomal recessive cerebellar ataxia with late-onset spasticity Autosomal recessive congenital cerebellar ataxia due to MGLUR1 deficiency Autosomal recessive congenital cerebellar ataxia due to GRID2 deficiency Ataxia-telangiectasia Ataxia-oculomotor apraxia type 1 Spinocerebellar ataxia with axonal neuropathy type 2 Spinocerebellar ataxia with axonal neuropathy type 1 Xeroderma pigmentosum-Cockayne syndrome complex Ataxia-telangiectasia-like disorder Xeroderma pigmentosum RIDDLE syndrome Friedreich ataxia Early-onset cerebellar ataxia with retained tendon reflexes Infantile onset spinocerebellar ataxia Marinesco-Sjögren syndrome Congenital cataracts-facial dysmorphism-neuropathy syndrome Posterior column ataxia-retinitis pigmentosa syndrome Early-onset progressive encephalopathy-spastic ataxia-distal spinal muscular atrophy syndrome Autosomal recessive spinocerebellar ataxia-blindness-deafness syndrome , a.k.a. spinocerebellar ataxia, autosomal recessive 3 (SCAR3) Autosomal recessive cerebellar ataxia-saccadic intrusion syndrome Autosomal recessive cerebellar ataxia-psychomotor delay syndrome Ataxia-oculomotor apraxia type 4 Gemignani syndrome , a.k.a. spinocerebellar ataxia-amyotrophy-deafness syndrome Cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome Acute infantile liver failure-cerebellar ataxia-peripheral sensory motor neuropathy syndrome , aka spinocerebellar ataxia, autosomal recessive 21 (SCAR21) Autosomal recessive ataxia due to ubiquinone deficiency Adult-onset autosomal recessive cerebellar ataxia Childhood-onset autosomal recessive slowly progressive spinocerebellar ataxia Infantile-onset autosomal recessive nonprogressive cerebellar ataxia , a.k.a. spinocerebellar ataxia, autosomal recessive 6 (SCAR6) Spectrin-associated autosomal recessive cerebellar ataxia Autosomal recessive cerebellar ataxia-epilepsy-intellectual disability syndrome due to WWOX deficiency Autosomal recessive cerebellar ataxia-epilepsy-intellectual disability syndrome due to TUD deficiency Autosomal recessive cerebellar ataxia-epilepsy-intellectual disability syndrome due to RUBCN deficiency Autosomal recessive cerebellar ataxia due to STUB1 deficiency Index of articles associated with the same name This article includes a list of related items that share the same name (or similar names).
-
Kabuki Syndrome
Wikipedia
In 1981, the two doctors separately submitted articles on this new diagnosis to the Journal of Pediatrics . [5] [23] [24] Dr Niikawa coined the term ‘Kabuki syndrome’ (also known as Kabuki make-up syndrome or Niikawa–Kuroki syndrome) as a reference to traditional Japanese theatre which he respected greatly. ... Gallery [ edit ] Individuals with Kabuki syndrome References [ edit ] ^ "Kabuki Syndrome Gene Identified" . ... "Developmental outcome in Kabuki syndrome". American Journal of Medical Genetics. ... "Unmasking Kabuki syndrome". Clinical Genetics . 83 (3): 201–11. doi : 10.1111/cge.12051 . ... External links [ edit ] Classification D ICD - 10 : Q87.0 ICD - 9-CM : 759.89 OMIM : 147920 MeSH : C537705 DiseasesDB : 32161 External resources GeneReviews : Kabuki Syndrome GARD : kabuki-syndrome Orphanet : 2322 Wikimedia Commons has media related to Kabuki syndrome . v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions )KMT2D, KDM6A, RAP1B, RAP1A, KMT2A, KMT2B, MKKS, MACROD2, ANOS1, HTC2, HNRNPK, MAPK1, MAP2K7, TUBA3D, CHD7, TERF2IP, FZR1, ANKRD11, RABGEF1, TNFRSF13B, ZFPM2, POGZ, KDM1A, SEPTIN9, GH1, MBD2, MYH6, POMC, XIST, UTY, EPHB2, TUBA3C, SOX3, SET, RBPJ, ITGB7, BRAF

-
22q11.2 Duplication Syndrome
Wikipedia
22q11.2 duplication syndrome Specialty Medical genetics 22q11.2 duplication syndrome is a rare genetic disorder caused by a duplication of a segment at the end of chromosome 22 . ... This is in sharp distinction to 22q11 deletion syndrome where about 90% of cases are caused by mutations that occur de novo . ... "Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients" . ... (August 2005). "22q11.2 duplication syndrome: two new familial cases with some overlapping features with DiGeorge/velocardiofacial syndromes". ... External links [ edit ] Classification D OMIM : 608363 MeSH : C567224 C567224, C567224 Genetics Home Reference DECIPHER database entry for 22q11.2 duplication syndrome v t e Mutation Mechanisms of mutation Insertion Deletion Substitution Transversion Transition Mutation with respect to structure Point mutation Nonsense mutation Missense mutation Conservative mutation Silent mutation Frameshift mutation Dynamic mutation Large-scale mutation Chromosomal translocations Chromosomal inversions Mutation with respect to overall fitness Deleterious mutation Advantageous mutation Neutral mutation Nearly neutral mutation Synonymous mutation Nonsynonymous mutation v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22
-
Nephrogenic Diabetes Insipidus
Wikipedia
Osmotic [ edit ] Other causes of acquired NDI include: hypokalemia (low blood potassium), post-obstructive polyuria, sickle cell disease or trait, amyloidosis, Sjögren syndrome , renal cystic disease, Bartter syndrome , and various medications ( amphotericin B , orlistat , ifosfamide , ofloxacin , cidofovir , vaptans ). [ citation needed ] In addition to kidney and systemic disorders, nephrogenic DI can present itself as a side effect of some medications . ... External links [ edit ] Classification D ICD - 10 : N25.1 ICD - 9-CM : 588.1 OMIM : 304800 125800 MeSH : D018500 External resources MedlinePlus : 000511 GeneReviews : Nephrogenic Diabetes Insipidus v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia v t e Diseases of ion channels Calcium channel Voltage-gated CACNA1A Familial hemiplegic migraine 1 Episodic ataxia 2 Spinocerebellar ataxia type-6 CACNA1C Timothy syndrome Brugada syndrome 3 Long QT syndrome 8 CACNA1F Ocular albinism 2 CSNB2A CACNA1S Hypokalemic periodic paralysis 1 Thyrotoxic periodic paralysis 1 CACNB2 Brugada syndrome 4 Ligand gated RYR1 Malignant hyperthermia Central core disease RYR2 CPVT1 ARVD2 Sodium channel Voltage-gated SCN1A Familial hemiplegic migraine 3 GEFS+ 2 Febrile seizure 3A SCN1B Brugada syndrome 6 GEFS+ 1 SCN4A Hypokalemic periodic paralysis 2 Hyperkalemic periodic paralysis Paramyotonia congenita Potassium-aggravated myotonia SCN4B Long QT syndrome 10 SCN5A Brugada syndrome 1 Long QT syndrome 3 SCN9A Erythromelalgia Febrile seizure 3B Paroxysmal extreme pain disorder Congenital insensitivity to pain Constitutively active SCNN1B / SCNN1G Liddle's syndrome SCNN1A / SCNN1B / SCNN1G Pseudohypoaldosteronism 1AR Potassium channel Voltage-gated KCNA1 Episodic ataxia 1 KCNA5 Familial atrial fibrillation 7 KCNC3 Spinocerebellar ataxia type-13 KCNE1 Jervell and Lange-Nielsen syndrome Long QT syndrome 5 KCNE2 Long QT syndrome 6 KCNE3 Brugada syndrome 5 KCNH2 Short QT syndrome KCNQ1 Jervell and Lange-Nielsen syndrome Romano–Ward syndrome Short QT syndrome Long QT syndrome 1 Familial atrial fibrillation 3 KCNQ2 BFNS1 Inward-rectifier KCNJ1 Bartter syndrome 2 KCNJ2 Andersen–Tawil syndrome Long QT syndrome 7 Short QT syndrome KCNJ11 TNDM3 KCNJ18 Thyrotoxic periodic paralysis 2 Chloride channel CFTR Cystic fibrosis Congenital absence of the vas deferens CLCN1 Thomsen disease Myotonia congenita CLCN5 Dent's disease CLCN7 Osteopetrosis A2, B4 BEST1 Vitelliform macular dystrophy CLCNKB Bartter syndrome 3 TRP channel TRPC6 FSGS2 TRPML1 Mucolipidosis type IV Connexin GJA1 Oculodentodigital dysplasia Hallermann–Streiff syndrome Hypoplastic left heart syndrome GJB1 Charcot–Marie–Tooth disease X1 GJB2 Keratitis–ichthyosis–deafness syndrome Ichthyosis hystrix Bart–Pumphrey syndrome Vohwinkel syndrome ) GJB3 / GJB4 Erythrokeratodermia variabilis Progressive symmetric erythrokeratodermia GJB6 Clouston's hidrotic ectodermal dysplasia Porin AQP2 Nephrogenic diabetes insipidus 2 See also: ion channels v t e Cell surface receptor deficiencies G protein-coupled receptor (including hormone ) Class A TSHR ( Congenital hypothyroidism 1 ) LHCGR ( Luteinizing hormone insensitivity , Leydig cell hypoplasia , Male-limited precocious puberty ) FSHR ( Follicle-stimulating hormone insensitivity , XX gonadal dysgenesis ) GnRHR ( Gonadotropin-releasing hormone insensitivity ) EDNRB ( ABCD syndrome , Waardenburg syndrome 4a , Hirschsprung's disease 2 ) AVPR2 ( Nephrogenic diabetes insipidus 1 ) PTGER2 ( Aspirin-induced asthma ) Class B PTH1R ( Jansen's metaphyseal chondrodysplasia ) Class C CASR ( Familial hypocalciuric hypercalcemia ) Class F FZD4 ( Familial exudative vitreoretinopathy 1 ) Enzyme-linked receptor (including growth factor ) RTK ROR2 ( Robinow syndrome ) FGFR1 ( Pfeiffer syndrome , KAL2 Kallmann syndrome ) FGFR2 ( Apert syndrome , Antley–Bixler syndrome , Pfeiffer syndrome , Crouzon syndrome , Jackson–Weiss syndrome ) FGFR3 ( Achondroplasia , Hypochondroplasia , Thanatophoric dysplasia , Muenke syndrome ) INSR ( Donohue syndrome Rabson–Mendenhall syndrome ) NTRK1 ( Congenital insensitivity to pain with anhidrosis ) KIT ( KIT Piebaldism , Gastrointestinal stromal tumor ) STPK AMHR2 ( Persistent Müllerian duct syndrome II ) TGF beta receptors : Endoglin / Alk-1 / SMAD4 ( Hereditary hemorrhagic telangiectasia ) TGFBR1 / TGFBR2 ( Loeys–Dietz syndrome ) GC GUCY2D ( Leber's congenital amaurosis 1 ) JAK-STAT Type I cytokine receptor : GH ( Laron syndrome ) CSF2RA ( Surfactant metabolism dysfunction 4 ) MPL ( Congenital amegakaryocytic thrombocytopenia ) TNF receptor TNFRSF1A ( TNF receptor associated periodic syndrome ) TNFRSF13B ( Selective immunoglobulin A deficiency 2 ) TNFRSF5 ( Hyper-IgM syndrome type 3 ) TNFRSF13C ( CVID4 ) TNFRSF13B ( CVID2 ) TNFRSF6 ( Autoimmune lymphoproliferative syndrome 1A ) Lipid receptor LRP : LRP2 ( Donnai–Barrow syndrome ) LRP4 ( Cenani–Lenz syndactylism ) LRP5 ( Worth syndrome , Familial exudative vitreoretinopathy 4 , Osteopetrosis 1 ) LDLR ( LDLR Familial hypercholesterolemia ) Other/ungrouped Immunoglobulin superfamily : AGM3, 6 Integrin : LAD1 Glanzmann's thrombasthenia Junctional epidermolysis bullosa with pyloric atresia EDAR ( EDAR hypohidrotic ectodermal dysplasia ) PTCH1 ( Nevoid basal-cell carcinoma syndrome ) BMPR1A ( BMPR1A juvenile polyposis syndrome ) IL2RG ( X-linked severe combined immunodeficiency ) See also cell surface receptorsAQP2, AVPR2, PRKCA, CLCNKB, AQP3, GRN, SIRT1, SLC4A4, RNF40, AVP, BBS1, CCDC28B, ARHGAP4, L1CAM, GPRC6A, SCT, VWF, FZD4, KEAP1, LGR6, LPAR3, VN1R17P, OXER1, MRGPRX3, MRGPRX1, GPR151, MRGPRX4, GPR166P, AQP1, ARHGAP1, COX8A, LNPEP, NFE2L2, ADRA2B, AQP8, AQP5, GPBAR1, BRS3, CALCA, MINDY4, CANX, CLCNKA, PDZD4, ACKR3, DMD, WDTC1, CXCR6, EDNRA, ELF3, LPAR2, EPHA3, G6PD, GABPA, ST14, SSTR4, SLC12A1, GPR42, REN, ADRA1A, ADCY6







