Research suggests that patients who are treated with ICIs are more susceptible to CNS disease (since the mechanism of ICIs induces adverse effects on the CNS due to augmented immune responses and neurotoxicity ). [28] The purpose of this exploration was to shed light on immunotherapies and distinguishing between neurotoxicity and brain metastasis in the early stages of treatment.
"Cardiovascular anomalies in children and young adults with Ullrich-Turner syndrome-the erlangen experience" . Clinical Cardiology . 28 (2): 88–92. doi : 10.1002/clc.4960280209 .
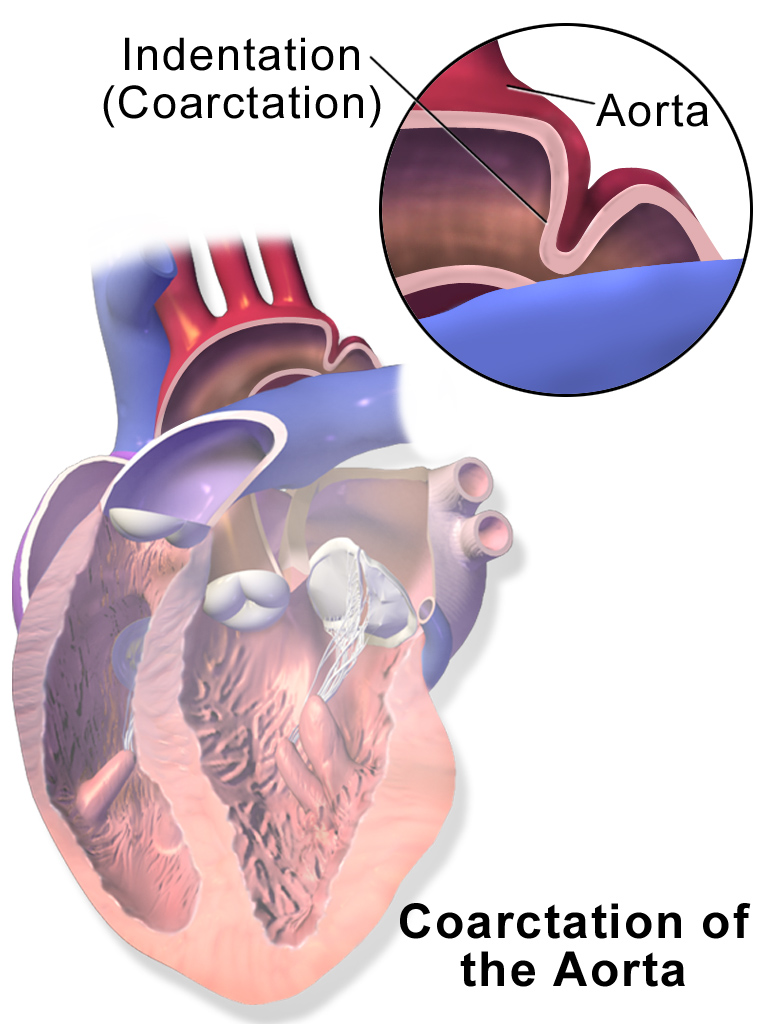
Overview The aorta is the largest artery in the body. It moves oxygen-rich blood from the heart to the rest of the body. Aortic coarctation (ko-ahrk-TAY-shun) is a narrowing of the aorta. It forces the heart to pump harder to move blood through the aorta. Coarctation of the aorta is generally present at birth (congenital heart defect). Symptoms can range from mild to severe. The condition might not be detected until adulthood. Coarctation of the aorta often occurs along with other congenital heart defects.
Critical congenital heart disease (CCHD) is a term that refers to a group of serious heart defects that are present from birth. These abnormalities result from problems with the formation of one or more parts of the heart during the early stages of embryonic development. CCHD prevents the heart from pumping blood effectively or reduces the amount of oxygen in the blood. As a result, organs and tissues throughout the body do not receive enough oxygen, which can lead to organ damage and life-threatening complications. Individuals with CCHD usually require surgery soon after birth. Although babies with CCHD may appear healthy for the first few hours or days of life, signs and symptoms soon become apparent.
Gough (1961) described the anomaly in father and son. He found 6 other reports of familial coarctation. In a systematic study of coarctation, Boon and Roberts (1976) discerned familial aggregation with multifactorial inheritance. Recurrence risks in sibs was about 0.5% for coarctation and 1.0% for any form of congenital heart defect. Beekman and Robinow (1985) described coarctation of the aorta in 4 generations. In 2 members of the family, mother and son, in the third and fourth generations, the coarctation was minimal; in the mother, for example, a gradient in the aorta was demonstrated mainly after peak exercise.
Archived from the original on 4 December 2014 . Retrieved 28 November 2014 . ^ Ziegelbauer, K; Speich, B; Mäusezahl, D; Bos, R; Keiser, J; Utzinger, J (Jan 2012).
However, separation also frequently occurs during labor and delivery, or with women carrying more than one baby. [28] Many cases of diastasis recti correct themselves after birth, but some do not.
In Nigeria, the use of imported Ready to Use Therapeutic Food (RUTF) has been used to combat malnutrition in the North, However, a research has shown that Soy Kunu , a locally sourced and prepared blend consisting of peanut, millet and soya beans contains the components of the Ready to Use Therapeutic Food (RUTF) and this has been used massively to reduce malnutrition in the north. [26] Breastfeeding [ edit ] Main article: Breastfeeding Breastfeeding can reduce rates of malnutrition and dehydration caused by diarrhea, but mothers are sometimes wrongly advised to not breastfeed their children. [10] Breastfeeding has been shown to reduce mortality in infants and young children. [12] Since only 38 percent of children worldwide under 6 months are exclusively breastfed, education programs could have large impacts on children malnutrition rates. [27] However, breastfeeding cannot fully prevent PEM if not enough nutrients are consumed. [5] Treatment [ edit ] Getting support to children with malnutrition in Kenya Treatment with antibiotics such as amoxicillin or cefdinir improve the response and survival rate of severely malnourished children to an outpatient treatment plan which provided therapeutic food . [2] This confirms the recommendation, "In addition to the provision of RUTF [ready-to-use therapeutic food], children need to receive a short course of basic oral medication to treat infections." contained in "Community-based management of severe acute malnutrition, A Joint Statement by the World Health Organization, the World Food Programme , the United Nations System Standing Committee on Nutrition and the United Nations Children’s Fund." [28] Epidemiology [ edit ] The World Health Organization estimates that malnutrition accounts for 54 percent of child mortality worldwide, [5] about 1 million children. [2] Another estimate also by WHO states that childhood underweight is the cause for about 35% of all deaths of children under the age of five years worldwide. [6] According to a 2008 review an estimated 178 million children under age 5 are stunted , most of whom live in sub-Saharan Africa. [12] A 2008 review of malnutrition found that about 55 million children are wasted, including 19 million who have severe wasting or severe acute malnutrition. [12] In 2020, a research paper that mapped stunting, wasting and underweight among children across 105 low- and middle-income countries found that only five countries were expected to meet global nutrition targets in all second administrative subdivisions . [29] As underweight children are more vulnerable to almost all infectious diseases, the indirect disease burden of malnutrition is estimated to be an order of magnitude higher than the disease burden of the direct effects of malnutrition. [6] The combination of direct and indirect deaths from malnutrition caused by unsafe water, sanitation and hygiene ( WASH ) practices is estimated to lead to 860,000 deaths per year in children under five years of age. [6] See also [ edit ] Child nutrition in Australia References [ edit ] ^ a b c d e f g Adam Wagstaff; Naoke Watanabe (November 1999).
Palmoplantar keratoderma (PPK) is a group of skin conditions characterized by thickening of the skin on the palms of the hands and soles of the feet. PPK can also be a feature of various underlying syndromes . In rare forms of PPK, organs other than the skin may also be affected. PPK can be either acquired during the lifetime (more commonly) or inherited. Acquired PPKs may arise due to changes in a person's health or environment. Inherited PPKs are caused by genetic mutations that result in abnormalities of keratin , a skin protein.
DPOAE suppression is significantly affected by age and becomes difficult to detect by approximately 50 years of age. [28] Spatial Hearing Advantage (dB) slowly increases through childhood and into adulthood.
The presiding judge, Masaharu Suda ( 須田 賢 , Suda Masaharu ) , sentenced Hachitani, then 33, to 12 months imprisonment, and Momii, then 39, for 18 months imprisonment, with both sentences suspended for 3 years. [25] The lawyers representing the controllers appealed, but the convictions were upheld on October 26, 2010 by the Supreme Court . [28] [29] In popular culture [ edit ] The events of the incident are documented in the final season 3 episode of the Discovery Channel documentary Aircrash Confidential . [30] The episode was first aired on 20 August 2018.
Nowadays it is completely accepted that the big majority of Mexico's and Latin America's mixed-race populations have the Mongolian spot [23] and that its presence works as an indicator of the actual degree of mestizaje present in a given population, [24] having its lower frequency in Uruguay with 36%, [24] followed by Argentina with an incidence 44%, [25] Mexico with 50%-52%, [26] 68% on Hispanic-Americans [27] and 88% on highland Peruvians. [28] A study performed in hospitals of Mexico City reported that, on average, 51.8% of Mexican newborns presented slate grey nevus, while it was absent on 48.2% of the analyzed babies. [25] According to the Mexican Social Security Institute nationwide, around half of Mexican babies have the slate grey nevus. [29] Central American indigenous children were subjected to racism due to their slate grey nevus but progressive circles began to make having the slate grey nevus popular after the late 1960s. [30] Highland Peruvians have the slate grey nevus. [31] Treatment [ edit ] As a congenital benign nevus , Mongolian spots do not require treatment and in most cases disappear before adolescence.
. ^ "An open letter to Melinda Gates" Archived 2007-09-28 at the Wayback Machine , Human Life International (2006-08-29). ^ Ssengooba, Freddie; et al. (2011).
Several surgical methods have been used, including: [26] Partial-thickness or full-thickness surgical blade excision Electrosurgery Curettage Laser surgery Cryosurgery Lesions removed by electrosurgery require an average of 30 to 33 days to heal, whereas lesions removed by surgical curettage require around 21 to 23 healing days. [27] During healing interval, the existing denture can be lined with a temporary tissue conditioner that acts as a palatal dressing and provides greater comfort. [26] Surgical removal of the lesion and the making of new dentures are effective in eradication of the lesion. [28] Good oral hygiene practice is very important in preventing repetition of events leading to the condition again. [29] Proper denture hygiene care should be carried out as instructed by your dentist and nocturnal use of dentures should be eliminated.
Sue Semple-Rowland at the University of Florida has recently restored sight in an avian model using gene therapy . [27] In March 2020, doctors at the Casey Eye Institute of the Oregon Health & Science University injected a CRISPR-modified virus into a patient's eye in an attempt to treat LCA10. [28] Popular culture [ edit ] In the episode "The Blackout in the Blizzard" (season 6, episode 16) of the television drama Bones , Dr.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-17 (LCA17) is caused by compound heterozygous mutation in the GDF6 gene (601147) on chromosome 8q22. For a general phenotypic description and a discussion of genetic heterogeneity of Leber congenital amaurosis (LCA), see LCA1 (204000). Clinical Features Asai-Coakwell et al. (2013) studied a female LCA patient with compound heterozygous mutations in the GDF6 gene who had vision limited to detection of hand motions, with an extinguished electroretinogram (ERG) typical of the LCA phenotype. She did not have other ocular or systemic phenotypes, but the authors noted that she had not undergone radiologic imaging to detect milder GDF6-induced skeletal disease. Evaluation of her clinically unaffected mother revealed a delayed rod b-wave implicit time on ERG; similarly, her apparently unaffected father showed reduced b-wave amplitude.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-12 (LCA12) is caused by homozygous mutation in the RD3 gene (180040) on chromosome 1q32. For a general phenotypic description and a discussion of genetic heterogeneity of LCA, see LCA1 (204000). Clinical Features Friedman et al. (2006) identified a sister and brother with Leber congenital amaurosis from a consanguineous Indian family. Both probands had had poor vision since birth. Nystagmus and atrophic lesions in the macular area with pigment migration were found on examination. Preising et al. (2012) reported a large consanguineous Kurdish family with LCA, in which 6 of 7 affected individuals were available for study.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-9 (LCA9) is caused by homozygous or compound heterozygous mutation in the NMNAT1 gene (608700) on chromosome 1p36. For a general discussion of the phenotypic and genetic heterogeneity in Leber congenital amaurosis, see LCA1 (204000). Description Early-onset neurodegeneration in the human retina can lead to Leber congenital amaurosis (LCA), the most severe human form of inherited photoreceptor-neuron degeneration resulting in congenital blindness, with an incidence of approximately 1 in 80,000 (summary by Koenekoop et al., 2012). NMNAT1 (608700) mutations consistently cause severe and rapidly progressive macular degeneration leading to severe central atrophy with an appearance of congenital macular coloboma in the neonatal period, as well as an unusual early-onset atrophy of the optic nerve (Perrault et al., 2012). Clinical Features Koenekoop et al. (2012) reexamined affected individuals from 8 families with Leber congenital amaurosis in whom they had identified mutations in the NMNAT1 gene (608700), which is ubiquitously expressed (see MOLECULAR GENETICS).
A number sign (#) is used with this entry because Leber congenital amaurosis-5 (LCA5) is caused by homozygous mutation in the gene encoding lebercilin (LCA5; 611408) on chromosome 6q14. For a general phenotypic description and a discussion of genetic heterogeneity of LCA, see LCA1 (204000). Clinical Features Dharmaraj et al. (2000) described a multigenerational kindred of Old Order River Brethren, a religious isolate descended from Swiss immigrants to America in the 1750s (Brechbill, 1972), segregating Leber congenital amaurosis. LCA in this kindred was not associated with multisystem abnormalities. Renal function remained normal. Neurologic and hepatic function were within normal limits.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-1 (LCA1) is caused by homozygous mutation in the gene encoding retinal guanylate cyclase (GUCY2D; 600179) on chromosome 17p13. Heterozygous mutation in the GUCY2D gene causes an allelic disorder, cone-rod dystrophy-6 (CORD6; 601777), and homozygous mutation in the same gene has also been found to cause autosomal recessive CORD (see 610777). Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009).
Leber congenital amaurosis (LCA) is an eye disorder that primarily affects the retina . People with this condition typically have severe visual impairment beginning in infancy. Other features include photophobia , involuntary movements of the eyes (nystagmus), and extreme farsightedness. The pupils also do not react normally to light. Additionally, the cornea may be cone-shaped and abnormally thin ( keratoconus ). Franceschetti's oculo-digital sign is characteristic of Leber congenital amaurosis.
A number sign (#) is used with this entry because Leber congenital amaurosis-11 (LCA11) is caused by heterozygous mutation in the IMPDH1 gene (146690) on chromosome 7q32. Heterozygous mutation in the IMPDH1 gene can also cause retinitis pigmentosa-10 (RP10; 180105). Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009).
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-16 (LCA16) is caused by homozygous mutation in the KCNJ13 gene (603208) on chromosome 2q37. For a general phenotypic description and a discussion of genetic heterogeneity of Leber congenital amaurosis, see LCA1 (204000). Clinical Features Sergouniotis et al. (2011) studied a consanguineous Middle Eastern family in which 2 brothers had nystagmus at birth and were diagnosed with Leber congenital amaurosis shortly thereafter. Poor night vision and difficulty reading print from an early age was reported for both patients; gradual progression of visual problems affecting central and peripheral vision was also noted. Both patients had bilateral cataract surgery in their third decade. Funduscopy revealed significant pigment in the retinal pigment epithelium (RPE), in a configuration unlike that of typical retinitis pigmentosa (see 268000).
A number sign (#) is used with this entry because Leber congenital amaurosis-7 can be caused by heterozygous or homozygous mutation in the CRX gene (602225) on chromosome 19q13. Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009). For a general description and a discussion of genetic heterogeneity of LCA, see 204000.
A number sign (#) is used with this entry because Leber congenital amaurosis-15 and juvenile retinitis pigmentosa are caused by homozygous or compound heterozygous mutation in the TULP1 gene (602280) on chromosome 6p21.3. Description Autosomal recessive childhood-onset severe retinal dystrophy is a heterogeneous group of disorders affecting rod and cone photoreceptors simultaneously. The most severe cases are termed Leber congenital amaurosis, whereas the less aggressive forms are usually considered juvenile retinitis pigmentosa (summary by Gu et al., 1997). Mutation in TULP1 can also cause a form of autosomal recessive retinitis pigmentosa (RP14; 600132). For a general phenotypic description and a discussion of the genetic heterogeneity of Leber congenital amaurosis, see LCA1 (204000); for retinitis pigmentosa, see 268000.
A number sign (#) is used with this entry because Leber congenital amaurosis-8 (LCA8) is caused by homozygous or compound heterozygous mutation in the CRB1 gene (604210) on chromosome 1q31. Homozygous or compound heterozygous mutation in CRB1 can also cause retinitis pigmentosa-12 (RP12; 600105). Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009).
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-6 (LCA6) is caused by homozygous or compound heterozygous mutation in the RPGRIP1 gene (605446) on chromosome 14q11. Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009). For a general description and a discussion of genetic heterogeneity of LCA, see 204000.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-2 (LCA2) is caused by homozygous or compound heterozygous mutation in the RPE65 gene (180069) on chromosome 1p31. Mutations in this gene also cause retinitis pigmentosa (RP20; 613794). Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009).
Leber congenital amaurosis (LCA) is a retinal dystrophy defined by blindness and responses to electrophysiological stimulation (Ganzfeld electroretinogram (ERG)) below threshold, associated with severe visual impairment within the first year of life. Epidemiology The prevalence of LCA is 1/50,000 - 1/33,000 live births and accounts for 5% of all retinal dystrophies and 20% of blindness in school age children. Clinical description LCA is characterized by severely reduced visual acuity (less or equal 20/400) or blindness within in the first year of life. Sluggish pupillary responses, roving eye movement, photophobia, high hyperopia, nystagmus, convergent strabismus, or keratoconus may occur depending on the genetic cause. The Franceschetti's oculo-digital sign, comprising eye poking, pressing, and rubbing is pathognomonic.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-10 (LCA10) is caused by homozygous or compound heterozygous mutations in the CEP290 gene (610142) on chromosome 12q21. Description Leber congenital amaurosis is a severe retinal dystrophy, causing blindness or severe visual impairment at birth or during the first months of life (summary by den Hollander et al., 2006). For a general phenotypic description and a discussion of genetic heterogeneity of Leber congenital amaurosis, see LCA1 (204000). Clinical Features Den Hollander et al. (2006) reported a consanguineous French Canadian family in which 4 sibs had Leber congenital amaurosis. The sibs were blind or severely visually impaired at birth. Two of the 4 experienced seizures but had no other neurologic symptoms.
Sorsby and Williams (1960) observed a family with multiple cases of retinal aplasia in which inheritance was autosomal dominant. 'Retinal aplasia' is the British term for what is called 'congenital amaurosis' on the continent. Much genetic heterogeneity exists as evidenced by the demonstration of both autosomal dominant and autosomal recessive forms. This disorder, which might be called an autosomal dominant form of Leber amaurosis congenita, must be very rare. Heckenlively (1988) reported a 6-generation family with a severe progressive retinal degeneration beginning in infancy.
Other areas more susceptible to fractures are the cribriform plate , the roof of orbits in the anterior cranial fossa , and the areas between the mastoid and dural sinuses in the posterior cranial fossa . [26] Prognosis [ edit ] Children with a simple skull fracture without other concerns are at low risk of a bad outcome and rarely require aggressive treatment. [27] The presence of a concussion or skull fracture in people after trauma without intracranial hemorrhage or focal neurologic deficits was indicated in long term cognitive impairments and emotional lability at nearly double the rate as those patients without either complication. [28] Those with a skull fracture were shown to have "neuropsychological dysfunction, even in the absence of intracranial pathology or more severe disturbance of consciousness on the GCS". [29] See also [ edit ] Le Fort facial fracture Facial fracture Mandibular fracture References [ edit ] ^ Haar FL (October 1975).
SSRI/SNRI withdrawal syndrome ) [22] [23] [24] blood pressure medications, including beta blockers such as propanolol and alpha-adrenergic agonists such as clonidine [25] [26] androgenic-anabolic steroids [27] [28] glucocorticoids [29] Rebound syndrome [ edit ] Main article: Rebound effect A wide range of drugs whilst not causing a true physical dependence can still cause withdrawal symptoms or rebound effects during dosage reduction or especially abrupt or rapid withdrawal. [30] [31] These can include caffeine , [32] stimulants, [33] [34] [35] [36] steroidal drugs and antiparkinsonian drugs. [37] It is debated whether the entire antipsychotic drug class causes true physical dependency, a subset, or if none do. [38] But, if discontinued too rapidly, it could cause an acute withdrawal syndrome. [39] When talking about illicit drugs rebound withdrawal, especially with stimulants, it is sometimes referred to as "coming down" or "crashing".