Fonknechten et al. (2000) analyzed DNA from 87 unrelated patients with autosomal dominant hereditary spastic paraplegia and detected 34 novel mutations scattered along the coding region of the SPG4 gene. They found missense (28%), nonsense (15%), and splice site point (26.5%) mutations as well as deletions (23%) and insertions (7.5%).
Spastic paraplegia type 4 (also known as SPG4) is the most common of a group of genetic disorders known as hereditary spastic paraplegias. These disorders are characterized by progressive muscle stiffness (spasticity) in the legs and difficulty walking. Hereditary spastic paraplegias are divided into two types: pure and complex. The pure types generally involve only spasticity of the lower limbs and walking difficulties. The complex types involve more widespread problems with the nervous system; the structure or functioning of the brain; and the nerves connecting the brain and spinal cord to muscles and sensory cells that detect sensations such as touch, pain, heat, and sound (the peripheral nervous system).
Spastic paraplegia 4 (SPG4) is the most common type of hereditary spastic paraplegia (HSP) inherited in an autosomal dominant manner. Disease onset ranges from infancy to older adulthood. SPG4 is characterized by slowly progressive muscle weakness and spasticity (stiff or rigid muscles) in the lower half of the body. In rare cases, individuals may have a more complex form with seizures, ataxia, and dementia. SPG4 is caused by mutations in the SPAST gene. Severity of symptoms usually worsens over time, however some individuals remain mildly affected throughout their lives. Medications, such as antispastic drugs and physical therapy may aid in stretching spastic muscles and preventing contractures (fixed tightening of muscles).
A rare form of hereditary spastic paraplegia with high intrafamilial clinical variability, characterized in most cases as a pure phenotype with an adult onset (mainly the 3rd to 5th decade of life, but that can present at any age) of progressive gait impairment due to bilateral lower-limb spasticity and weakness as well as very mild proximal weakness and urinary urgency. In some cases, a complex phenotype is also reported with additional manifestations including cognitive impairment, cerebellar ataxia, epilepsy and neuropathy. A faster disease progression is noted in patients with a later age of onset.
Association With HIV-1 Disease Progression Carrington et al. (1999) reported that the extended survival of 28 to 40% of HIV-1-infected Caucasian patients who avoided AIDS for 10 or more years (see 609423) could be attributed to their being fully heterozygous at HLA class I loci, to lacking the AIDS-associated alleles B*35 and Cw*04, or to both.
Recombinant events narrowed the critical region to an approximately 28-cM interval, between markers D6S251 and D6S468, a region overlapping that previously assigned to MCDR1.
North Carolina macular dystrophy (NCMD) is an inherited eye disorder that affects the development of the macula , the small, but important part of the eye located in the center of the retina . The macula allows a person to see fine details and do tasks that require central vision, such as reading and driving. It is also important for seeing colors. The severity of changes in the development of the macula varies, causing some people to have little or no vision loss, while others may have severe vision loss. NCMD is considered non-progressive, which means most researchers believe the vision loss does not change after birth. Others believe it may progress slowly through age twelve.[14914] However, vision loss may increase if complications develop, such as new, abnormal blood vessels growing under the retina (choroidal neovascularization).
North Carolina macular dystrophy (NCMD) is a non-progressive autosomal dominant macular disorder of congenital or infantile onset characterized by loss of central vision, the accumulation of drusen in the macula and atrophy of photoreceptor cells with a variable phenotype at macular examination.
Immunoreactive ET1 was detected in 2 cultured media. Hypoxia treatment for 28 hours increased immunoreactive ET1 levels approximately 1.3-fold in 1 cultured cell medium but decreased it in 2 cell lines.
Also so called XYY syndrome can often cause speech delay. [25] Neonatal Brachial Plexus Palsy - There exists a high prevalence of early language delay exists among toddlers with neonatal brachial plexus palsy. [26] Poverty - Poverty is a risk factor for language delay as a result of a lack of access appropriate therapies. [27] Socioeconomic adversities correlate with delayed language development. [28] Psychosocial deprivation - The child doesn't spend enough time communicating with adults, such as babbling and joint attention.
A seasonal pattern is seen in outbreaks, with a marked increase in cases reported in the late summer and early fall. [23] The CDC has determined and submitted to GenBank complete or nearly complete genomic sequences for three known strains of the virus, which are "genetically related to strains of EV-D68 virus that were detected in previous years in the United States, Europe, and Asia." [24] While rates of paralytic symptoms appear to be correlated with the number of respiratory infections, in initial anecdotal reports, the cases are not clustered within a family or school, suggesting that the paralysis per se is not directly contagious, but arises as a very rare complication of the common respiratory infection. [20] Cases of similar illnesses have been reported in Canada, Northern Europe, and Japan. [18] Over 90% of reported cases are in children. [25] History [ edit ] AFM has only been formally tracked since 2014, since the incidence has spiked in recent years. [ citation needed ] A group in Texas reported having observed a pattern in 2013 of one to four cases per year with similar polio-like characteristics. [20] In 2014, the CDC Morbidity and Mortality Weekly Report [14] and a CDC Clinician Outreach and Communication Activity (COCA) conference call, [26] noted that many cases had neck, back, or extremity pain, but otherwise those affected generally had normal sensation in their limbs. [27] A few participants in the conference call discussed whether pain, later abating, might precede the onset of paralysis. [26] [28] An October 2014 report described outbreaks in California and Colorado, suggesting that the number of cases might be 100 or more nationwide. [29] Diagnosis included a detailed medical history , MRI imaging, and the elimination of transverse myelitis or Guillain–Barré syndrome as potential causes.
Acute flaccid myelitis (AFM) is a condition that affects the spinal cord leading to muscle weakness and loss of reflexes. Most people who develop AFM have had a viral illness with flu-like symptoms one to four weeks before symptoms of AFM. Symptoms of AFM include sudden onset (acute) of weakness in the arm(s) or leg(s), loss of muscle tone, and decreased or absent reflexes. Other symptoms may include pain, facial weakness, and difficulty swallowing, speaking, or moving the eyes. It is not clear why some people develop AFM and others do not. Diagnosis is based on the symptoms, a clinical exam, an MRI of the spine, and other laboratory testing.
Outcome was shown to be better in people with higher IQ, [27] social status, [28] greater educational attainments, [29] younger age of onset and diagnosis, [29] attacks with less dramatic features, [29] and fewer additional somatoform complaints. [29] Epidemiology [ edit ] The number of people with PNES ranges from 2 to 33 per 100,000. [30] Although the rate in the general population are difficult to determine, it remains the most frequent non-epileptic condition seen in epilepsy centers.
Recurrent respiratory papillomatosis is a rare respiratory disease characterized by the development of exophytic papillomas, affecting the mucosa of the upper aero-digestive tract (with a strong predilection for the larynx), caused by an infection with human papilloma virus. Symptoms at presentation may include hoarseness, chronic cough, dyspnea, recurrent upper respiratory tract infections, pneumonia, dysphagia, stridor, and/or failure to thrive. Epidemiology The prevalence of recurrent respiratory papillomatosis (RRP) is estimated at about 1/70,400 in the United Kingdom. Annual incidence of the disease is about 1/23,300 in children and 1/55,500 in adults in the United States. The adult form affects males more often than females. Clinical description A bimodal age distribution is characteristic, with the disease affecting either young children or young adults.
Recurrent respiratory papillomatosis (RRP) is a rare viral disease where tumors (papillomas) grow in the air passages leading from the nose and mouth into the lungs (respiratory tract). There are two types, a juvenile-onset form and an adult-onset form. The tumors can cause a hoarse voice, chronic cough, and difficulty breathing. They may vary in size and grow very quickly, and may grow back even when removed. These tumors rarely become cancerous, but can cause long-term airway and voice complications. RRP is caused by two types of human papillomavirus (HPV) , called HPV 6 and HPV 11.
and sends him to work in a neighborhood clinic, instead. [27] [28] Q fever was also highlighted in an episode of the U.S. television medical drama House (" The Dig ", season seven, episode 18).
Q fever, caused by Coxiella burnetii , is a bacterial zoonosis with a wide clinical spectrum that can be life-threatening and, in some cases, can become chronic. Epidemiology In recent years in Europe, there has been an increase in the number of reported cases. In the Netherlands in particular, between 2007 and 2010 there were more than 4,000 cases reported. Clinical description Q fever affects all ages, but is mostly reported in those aged 30-70 years. The incubation period is 2-3 weeks. Approximately 60% of cases are asymptomatic.
Q fever is a worldwide disease with acute and chronic stages caused by the bacteria known as Coxiella burnetii . Cattle, sheep, and goats are the primary reservoirs although a variety of species may be infected. Organisms are excreted in birth fluids, milk, urine, and feces of infected animals and are able to survive for long periods in the environment. Infection of humans usually occurs by inhalation of these organisms from air that contains airborne barnyard dust contaminated by dried placental material, birth fluids, and waste products of infected animals. Other modes of transmission to humans, including tick bites, ingestion of unpasteurized milk or dairy products, and human to human transmission, are rare.
"Decreased subunit stability as a novel mechanism for potassium current impairment by a KCNQ2 C terminus mutation causing benign familial neonatal convulsions" . J Biol Chem . 281 (1): 418–28. doi : 10.1074/jbc.M510980200 . PMID 16260777 . ^ a b c Claes L, Ceulemans B, Audenaert D, Deprez L, Jansen A, Hasaerts D, Weckx S, Claeys K, Del-Favero J, Van Broeckhoven C, De Jonghe P (2004).
A number sign (#) is used with this entry because benign familial infantile seizures-2 (BFIS2) is caused by heterozygous mutation in the PRRT2 gene (614386) on chromosome 16p11. Description Benign familial infantile seizure is an autosomal dominant disorder characterized by afebrile partial complex or generalized tonic-clonic seizures occurring between 3 and 12 months of age with a good response to medication and no neurologic sequelae. Seizures usually remit by age 18 months (summary by Weber et al., 2004). For a general phenotypic description and a discussion of genetic heterogeneity of benign familial infantile seizures, see BFIS1 (601764). Benign familial infantile seizures can also occur in 2 allelic disorders: infantile convulsions and choreoathetosis (ICCA; 602066) and paroxysmal kinesigenic choreoathetosis (EKD1; 128200).
Description Benign familial infantile seizures (BFIS) is a seizure disorder of early childhood with age at onset from 3 months up to 24 months. It is characterized by brief seizures beginning with slow deviation of the head and eyes to 1 side and progressing to generalized motor arrest and hypotonia, apnea and cyanosis, and limb jerks. Seizures usually occur in clusters over a day or several days. The ictal EEG shows focal parietal-temporal activity, whereas the interictal EEG is normal. Concurrent and subsequent psychomotor and neurologic development are normal (Franzoni et al., 2005). See also benign familial neonatal seizures (BFNS1; 121200). Deprez et al. (2009) provided a review of the genetics of epilepsy syndromes starting in the first year of life, and included a diagnostic algorithm.
For a phenotypic description and a discussion of genetic heterogeneity of benign neonatal seizures, see BFNS1 (121200). Clinical Features Schiffmann et al. (1991) described an Iranian Jewish kindred in which 4 children of the same generation in 2 separate sibships with complex parental consanguinity had neonatal seizures. Linkage analysis excluded assignment to the BFNS1 locus on chromosome 20q. Inheritance The transmission pattern of benign neonatal seizures in the family reported by Schiffmann et al. (1991) suggested autosomal recessive inheritance. INHERITANCE - Autosomal recessive NEUROLOGIC Central Nervous System - Seizures, generalized tonic-clonic - Hypertonia in neonatal period - Normal interictal EEG - Normal psychomotor development - Patients may develop a seizure disorder later in life MISCELLANEOUS - Onset at 6-36 hours of life - Seizures resolve by 4 months of age - See EBN1 ( 121200 ) for an autosomal dominant form ▲ Close
A number sign (#) is used with this entry because of evidence that benign familial neonatal seizures-1 (BFNS1) is caused by heterozygous mutation in the KCNQ2 gene (602235) on chromosome 20q13. Some patients with KCNQ2 mutations develop severe early infantile epileptic encephalopathy (EIEE7; 613720). Description Benign familial neonatal seizures is an autosomal dominant disorder characterized by clusters of seizures occurring in the first days of life. Most patients have spontaneous remission by 12 months of age. The disorder is distinguished from benign familial infantile seizures (BFIS1; 601764) by an earlier age at onset. Deprez et al. (2009) provided a review of the genetics of epilepsy syndromes starting in the first year of life, and included a diagnostic algorithm.
A number sign (#) is used with this entry because benign familial neonatal seizures-2 (BFNS2) is caused by heterozygous mutation in the KCNQ3 gene (602232) on chromosome 8q24. Description Benign familial neonatal seizures-2 is an autosomal dominant neurologic condition characterized by onset of clonic or tonic-clonic seizures in the first few days of life. Seizures tend to last for about a minute, may occur several times a day, and are responsive to medication. Almost all patients have full remission within the first months of life, although some rare patients may have a few seizures later in childhood. EEG, brain imaging, and psychomotor development are usually normal (summary by Fister et al., 2013).
Benign familial neonatal-infantile seizures (BFNIS) is a benign familial epilepsy syndrome with an intermediate phenotype between benign familial neonatal seizures (BFNS) and benign familial infantile seizures (BFIS; see these terms). So far, this syndrome has been described in multiple members of 10 families. Age of onset in these BFNIS families varied from 2 days to 6 months, with spontaneous resolution in most cases before the age of 12 months. Like BFNS and BFIS, seizures in BFNIS generally occur in clusters over one or a few days with posterior focal seizure onset. BFNIS is caused by mutations in the SCN2A gene (2q24.3), encoding the voltage-gated sodium channel alpha-subunit Na(V)1.2.
For a phenotypic description and a discussion of genetic heterogeneity of benign familial neonatal seizures, see BFNS1 (121200). Clinical Features Concolino et al. (2002) reported a family in which 3 members over 3 generations had benign neonatal seizures inherited in an autosomal dominant pattern. Cytogenetics By cytogenic analysis in a family with benign neonatal seizures, Concolino et al. (2002) identified a pericentric inversion of chromosome 5, inv(5)(p15q11), which was present in all 3 affected members and absent in 3 unaffected first-degree relatives. The authors noted that the breakpoint was different from that found in cri-du-chat syndrome (123450). INHERITANCE - Autosomal dominant NEUROLOGIC Central Nervous System - Seizures, generalized tonic-clonic MISCELLANEOUS - Onset in first months of life - Seizures usually remit spontaneously by 12 months of age - No neurologic sequelae ▲ Close
For a general phenotypic description and a discussion of genetic heterogeneity of benign familial infantile seizures, see BFIS1 (601764). Clinical Features Li et al. (2008) reported a 4-generation Chinese family in which 8 individuals had benign infantile seizures. Age at seizure onset ranged from 3 to 10 months of life. The proband developed afebrile seizures at age 6 months, characterized by staring, eye deviation, focal clonus of the face, followed by secondarily generalized phase with hypertonia, extremities clonus, cyanosis, and urinary incontinence. The seizures lasted from 30 seconds to 2 minutes and occurred mainly in clusters with 3 to 10 episodes per day. Interictal EEG, brain CT, and MRI were classified as normal. Treatment with sodium valproate was effective, and the proband's seizures spontaneously remitted by 3 years of age.
Benign familial infantile epilepsy (BFIE) is a genetic epileptic syndrome characterized by the occurrence of afebrile repeated seizures in healthy infants, between the third and eighth month of life. Epidemiology Although BFIE cases have been reported worldwide, prevalence and incidence remain unknown. In an Argentinian case series, BFIE have been listed as the third most common type of epilepsy in the first two years of life. Clinical description Seizures usually occur between 3 to 8 months of life, with clusters (8-10 a day) of repeated and brief episodes (2-5 minutes) over a few days. They are usually focal but can sometimes become generalized. Patients present with motor arrest, unresponsiveness, head and/or eye deviation to one side, staring, fluttering of eyelids, grunting, cyanosis, diffuse hypertonia and unilateral or bilateral clonic jerks of the limbs.
A number sign (#) is used with this entry because of evidence that benign familial infantile seizures-5 (BFIS5) is caused by heterozygous mutation in the SCN8A gene (600702) on chromosome 12q13. Heterozygous mutation in the SCN8A gene can also cause the more severe disorder EIEE13 (614558). Description Benign familial infantile seizures-5 (BFIS5) is an autosomal dominant neurologic disorder characterized by onset of afebrile seizures during infancy. In most cases, the seizures remit by age 2 years, although some patients may have single or a few seizures later in childhood. The seizures respond well to treatment with sodium channel blockers, and patients have normal subsequent psychomotor development.
Benign familial neonatal epilepsy (BFNE) is a rare genetic epilepsy syndrome characterized by the occurrence of afebrile seizures in otherwise healthy newborns with onset in the first few days of life. Epidemiology Prevalence is currently unknown since this disorder is possibly overlooked. About 100 families have been reported to date. Clinical description Seizure onset is usually between the second and the eighth day of life, in otherwise healthy newborns. Seizures are mostly focal involving alternatively both sides of the body and apnea is frequently associated. Seizures can be isolated or in clusters, are generally brief and last 1-2 minutes.
A number sign (#) is used with this entry because benign familial neonatal-infantile seizures-3 (BFIS3) is caused by heterozygous mutation in the SCN2A gene (182390) on chromosome 2q24. See also early infantile epileptic encephalopathy-11 (EIEE11; 613721), a more severe disorder that also results from mutations in the SCN2A gene. Description Benign familial neonatal-infantile seizures is an autosomal dominant disorder in which afebrile seizures occur in clusters during the first year of life, without neurologic sequelae (Shevell et al., 1986). For a general phenotypic description and a discussion of genetic heterogeneity of benign familial infantile seizures, see BFIS1 (601764). BFIS1 has a slightly later onset than BFIS3, while benign neonatal seizures (see BFNS1, 121200) has a slightly earlier onset.
Lawrence River Valley is the site of the most frequent infections, with 20–30% of the population testing positive. [25] A review of reported cases in 2018 showed disease presence throughout Southeast Asia , [26] In India, the Gangetic West Bengal is the site of most frequent infections, with 9.4% of the population testing positive. [27] H. c. capsulatum was isolated from the local soil proving endemicity of histoplasmosis in West Bengal. [28] History [ edit ] Histoplasma was discovered in 1905 by Samuel T.
Overview Histoplasmosis is an infection caused by breathing in spores of a fungus often found in bird and bat droppings. People usually get it from breathing in these spores when they become airborne during demolition or cleanup projects. Soil contaminated by bird or bat droppings also can spread histoplasmosis, putting farmers and landscapers at a higher risk of the disease. In the United States, histoplasmosis commonly occurs in the Mississippi and Ohio River valleys. But it can occur in other areas, too. It also occurs in Africa, Asia, Australia, and in parts of Central and South America.
A rare mycosis characterized by granulomatous inflammation primarily of the lung after inhalation of spores of Histoplasma capsulatum . The severity of clinical disease depends on the immune status of the individual and the size of the inoculum. In immunocompetent persons, the infection usually takes a self-limiting and asymptomatic or relatively mild, flu-like course. In immunocompromised patients, it can become progressive and disseminated, involving multiple organs and presenting with fever, pneumonia, hepatosplenomegaly, skin infiltrates, and endocarditis, among others.
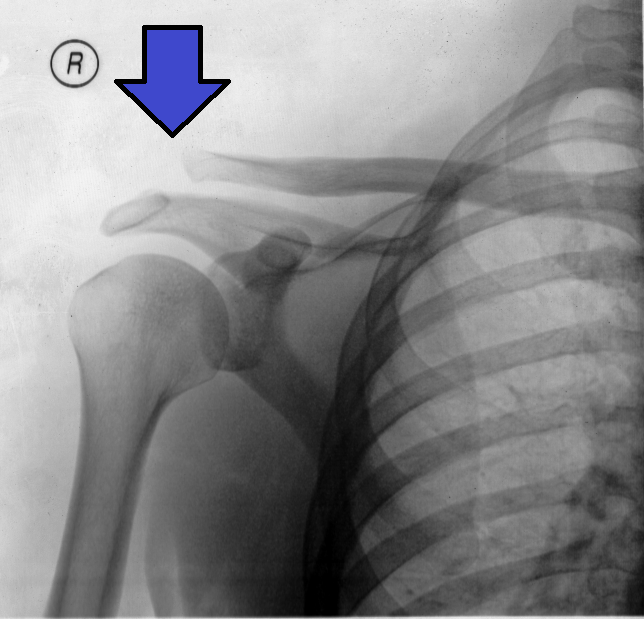
They found that 67% (413 patients) of the patients improved, while 28% did not improve and went to surgical treatment. 5% did not improve and declined further treatment. [ citation needed ] Of the 413 patients who improved, 74 had a recurrence of symptoms during the observation period and their symptoms responded to rest or after resumption of the exercise program. [ citation needed ] The Morrison study shows that the outcome of impingement symptoms varies with patient characteristics.
Atrioventricular nodal reentry tachycardia (AVNRT) Atrioventricular nodal reentry tachycardia (AVNRT) is the most common type of supraventricular tachycardia. People with AVNRT have episodes of an irregularly fast heartbeat (more than 100 beats per minute) that often start and end suddenly. The episodes are due to an extra pathway — called a reentrant circuit — located in or near the AV node that causes the heart to beat prematurely. AVNRT tends to occur more often in young women, but it can affect anyone. Diagnosis Tests and procedures used to diagnose AVNRT may include: Blood tests to check thyroid function, heart disease or other conditions that may trigger an irregular heartbeat Electrocardiogram (ECG) to measure the electrical activity of the heart and measure the timing and duration of each heartbeat Holter monitor , which is a portable ECG device designed to record the heart's activity as you perform everyday activities Echocardiogram, which uses sound waves to produce images of the heart's size, structure and motion Your health care provider might also try to trigger an episode with other tests, which may include: Stress test, which is typically done on a treadmill or stationary bicycle while the heart activity is monitored Electrophysiological study and cardiac mapping, which can reveal the precise location of the irregular heartbeat (arrhythmia) Treatment Most people with AVNRT don't need medical treatment.
Furthermore, the ADHD-C and ADHD-PI presenting did not differ on measures of sustained attention." [28] Epidemiology [ edit ] It is difficult to say exactly how many children or adults worldwide have ADHD because different countries have used different ways of diagnosing it, while some do not diagnose it at all.
"Median cheilitis" may be seen, which is fissuring in the midline of the lips due to the enlargement of the lips. [28] Angular cheilitis may also be associated with orofacial granulomatosis.
Because this conversion in endosomal membranes changes dynamically with fission and fusion events to create/absorb intracellular transport vesicles , enlarged cytoplasmic vacuoles have been found in patient neurons, muscle, and cartilage. [25] [26] These have been identified as intracytoplasmic vacuoles(fluid sacs inside cellular cytoplasm ) causing excessive build-up of vacuolated macrophages in bone marrow and pericardial fluid in the heart. [27] [28] Fluids may also accumulate in a choroid spaces under the retina, causing central serous retinopathy or chorioretinopathy and possibly vision loss. [29] Paradoxically, overexpression of FIG4 does not yield obvious morphologic phenotype of these fluids accumulating, but alters PI(3,5)P2 levels making cells prone to expansion through dilation of intracellular membranes .
Yunis-Varon syndrome is a rare condition that affects many different parts of the body. Signs and symptoms are generally present from birth and may include underdeveloped or absent collarbones (clavicles); large fontanelles ; characteristic facial features; hypotonia (reduced muscle tone) and/or abnormalities of the fingers and toes. Affected people may also experience feeding difficulties, breathing problems, brain malformations, heart defects , skeletal abnormalities, developmental delay, and/or intellectual disability. Yunis-Varon syndrome is caused by changes (mutations) in the FIG4 gene and is inherited in an autosomal recessive manner. Treatment is based on the signs and symptoms present in each person.
A number sign (#) is used with this entry because Yunis-Varon syndrome (YVS) is caused by homozygous or compound heterozygous mutation in the FIG4 gene (609390) on chromosome 6q21. Description Yunis-Varon syndrome is a severe autosomal recessive disorder characterized by skeletal defects, including cleidocranial dysplasia and digital anomalies, and severe neurologic involvement with neuronal loss. Enlarged cytoplasmic vacuoles are found in neurons, muscle, and cartilage. The disorder is usually lethal in infancy (summary by Campeau et al., 2013). Clinical Features Yunis and Varon (1980) described 5 children in 3 families with cleidocranial dysplasia (absent clavicles, macrocrania, diastasis of sutures), micrognathia, absent thumbs and distal phalanges of fingers, hypoplasia of proximal phalanx and absence of distal phalanx of the big toes, pelvic dysplasia, bilateral hip dislocation, and retracted and poorly delineated lips.
A rare, genetic, multiple congenital malformation syndrome, characterized by cleidocranial dysplasia (wide fontanelles, calvaria dysostosis, absent or hypoplastic clavicles), absent thumbs and halluces, hypoplastic distal and medial phalanges of fingers, pelvic dysplasia with hip dislocations. Dysmorphic features include sparse scalp hair, protruding eyes, low-set ears, anteverted nares, midfacial hypoplasia, tented upper lip, high arched palate, and micrognathia. Brain malformations are frequently associated. From birth, affected individuals tend to be significantly hypotonic and present with global developmental delay, and respiratory, feeding and swallowing difficulties.
Worry has also been hypothesized to contain more imagery than rumination; however, support for this has been mixed. [28] [29] [30] Overall, these studies suggest that worry and rumination are related constructs that both lead to depression and anxiety.
It is simply a fight for life." [27] Brazil carried out such a compulsory licensing threat for the first time in May 2007, on efavirenz , produced by Merck. [28] The agreements signed on November 14, 2001 at the WTO conference in Qatar reaffirmed that TRIPs "does not and should not prevent Members from taking measures to protect public health" including "medicines for all." [19] That same year, the United Nations Commission on Human Rights affirmed access to AIDS drugs as a human right unanimously, with the exception of the abstention of the United States .