Syndromic multisystem autoimmune disease due to Itch deficiency is a rare, genetic, systemic autoimmune disease characterized by failure to thrive, global developmental delay, distictive craniofacial dysmorphism (relative macrocephaly, dolichocephaly, frontal bossing, orbital proptosis, flattened midface with a prominent occiput, low, posteriorly rotated ears, micrognatia), hepato- and/or splenomegaly, and multisystemic autoimmune disease involving the lungs, liver, gut and/or thyroid gland.
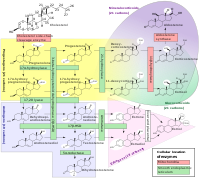
Mapping Lohr et al. (2010) performed genomewide autozygosity mapping in 5 Old Order Amish patients with syndromic multisystem autoimmune disease and identified a 19-Mb homozygous block in the pericentromeric region of chromosome 20, bounded by rs2038383 and rs2067084 and containing 258 known or hypothetical genes. ... Molecular Genetics In 10 Old Order Amish patients with syndromic multisystem autoimmune disease mapping to chromosome 20q11, Lohr et al. (2010) sequenced the candidate gene ITCH and identified homozygosity for a frameshift mutation (606409.0001) in all.
Overview Esophageal spasms are painful contractions within the muscular tube connecting your mouth and stomach. This tube is called the esophagus. Esophageal spasms can feel like sudden, severe chest pain that lasts from a few minutes to hours. Some people may mistake it for heart pain, also called angina. Esophageal spasms typically occur only occasionally and might not need treatment. But sometimes the spasms are frequent and can prevent food and liquids from traveling through the esophagus. If esophageal spasms interfere with your ability to eat or drink, treatments are available.
This article has multiple issues. Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article needs additional citations for verification . Please help improve this article by adding citations to reliable sources . Unsourced material may be challenged and removed. Find sources: "Esophageal spasm" – news · newspapers · books · scholar · JSTOR ( August 2019 ) ( Learn how and when to remove this template message ) Some of this article's listed sources may not be reliable . Please help this article by looking for better, more reliable sources. Unreliable citations may be challenged or deleted. ( August 2019 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) Esophageal spasm Other names Oesophageal spasm Specialty Gastroenterology Differential diagnosis Achalasia , angina , scleroderma , esophageal cancer , esophagitis [1] Esophageal spasm is a disorder of motility of the esophagus . [2] There are two types of esophageal spasm: [2] Diffuse or distal esophageal spasm (DES), where there is uncoordinated esophageal contractions Nutcracker esophagus (NE) also known as hypertensive peristalsis, where the contractions are coordinated but with an excessive amplitude.
Purine nucleoside phosphorylase (PNP) deficiency is a disorder of the immune system (primary immunodeficiency) characterized by recurrent infections, neurologic symptoms, and autoimmune disorders . PNP deficiency causes a shortage of white blood cells, called T-cells , that help fight infection. Some people with this condition develop neurologic symptoms, such as stiff or rigid muscles (spasticity), uncoordinated movements (ataxia), developmental delay, and intellectual disability. In addition, PNP deficiency is associated with an increased risk to develop autoimmune disorders, such as autoimmune hemolytic anemia , idiopathic thrombocytopenic purpura (ITP), autoimmune neutropenia , thyroiditis , and lupus . PNP deficiency is caused by mutations in the PNP gene and is inherited in an autosomal recessive manner.
Purine nucleoside phosphorylase deficiency is a disorder of the immune system called an immunodeficiency. Immunodeficiencies are conditions in which the immune system is not able to protect the body effectively from foreign invaders such as bacteria and viruses. People with purine nucleoside phosphorylase deficiency have low numbers of immune system cells called T cells , which normally recognize and attack foreign invaders to prevent infection. Some affected individuals also have low numbers of other immune system cells called B cells, which normally help fight infections by producing immune proteins called antibodies (or immunoglobulins). These proteins target foreign invaders and mark them for destruction.
The metabolic abnormalities resembled those in the erythrocytes of patients with the Lesch-Nyhan syndrome (300322). The immune defect was thought to be related to inhibition of adenosine deaminase by inosine. ... Stephenson and Tolmie (1990) were prompted to restudy this family after diagnosing PNP deficiency in a young girl who presented with dysequilibrium syndrome with pyramidal signs, including extensor plantar responses and exaggerated reflexes but not prominent spasticity, very similar to the neurologic picture in the family reported by Graham-Pole et al. (1975).
A rare immune disease characterized by progressive immunodeficiency leading to recurrent and opportunistic infections, autoimmunity and malignancy as well as neurologic manifestations. Epidemiology To date, more than 70 patients have been reported with Purine nucleoside phosphorylase (PNP) deficiency in the world literature. PNP deficiency accounts for less than 4% of patients with severe combined immunodeficiency (SCID). Clinical description PNP deficiency typically manifests in the 1st years of life, with recurrent, opportunistic infections caused by bacterial, viral (such as varicella zoster virus), and fungal (such as Pneumocystis carinii ) pathogens. The susceptibility to infections is variable ranging from classical SCID in infancy to infrequent infections during childhood and even the 2nd decade of life.
PFAPA (Periodic fever - aphthous stomatitis- pharyngitis - adenopathy) syndrome is an auto inflammatory syndrome characterized by recurrent febrile episodes associated with aphthous stomatitis, pharyngitis and cervical adenitis. ... Differential diagnosis Differential diagnosis includes other diseases characterized by periodic fever such as recurrent tonsillitis, streptococcal infection, juvenile idiopathic arthritis, Behçet's disease, cyclic neutropenia, familial Mediterranean fever, TRAPS syndrome, and mevalonate kinase deficiency (see these terms).
A number sign (#) is used with this entry because of evidence that Van Esch-O'Driscoll syndrome (VEODS) is caused by hemizygous mutation in the POLA1 gene (312040) on chromosome Xp22. Description Van Esch-O'Driscoll syndrome (VEODS) is characterized by varying degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations (Van Esch et al., 2019). ... Van Esch et al. (2019) studied 9 patients from 5 families with syndromic X-linked mental retardation, including the Belgian family (family A) originally reported by Van Esch et al. (2005). ... Molecular Genetics In 9 affected male patients from 5 unrelated families with syndromic X-linked mental retardation, Van Esch et al. (2019) identified 5 different mutations in the POLA1 gene (312040.0002-312040.0006). ... Exclusion Studies In a 5-generation Belgian family with syndromic X-linked mental retardation mapping to Xp22.1-p21.3, Van Esch et al. (2005) screened 2 MRX-associated genes mapping within the 6-cM interval, ARX (300382) and IL1RAPL1 (300206), but detected no mutations.
A rare, genetic, syndromic intellectual disability characterized by developmental delay, mild to moderate intellectual disability, low birth weight, moderate to severe short stature, microcephaly and variable hypergonadotropic hypogonadism.
A number sign (#) is used with this entry because of evidence that X-linked syndromic mental retardation-11 (MRXS11) is caused by mutation in the RBMX gene (300199) on chromosome Xq26. ... Clinical Features In a large family from North Carolina, Shashi et al. (2000) studied a seemingly novel X-linked mental retardation syndrome with characteristic facial dysmorphic features. ... The blood-lymphocyte karyotype and the results of DNA analysis for fragile X syndrome (300624) and of other routine investigations were normal. ... Mapping By linkage analysis with polymorphic DNA markers spanning the X chromosome in a large family with an X-linked mental retardation syndrome, Shashi et al. (2000) established linkage to Xq26-q27. ... The diagnosis of Shashi X-linked mental retardation syndrome was suggested by the clinical features of coarse facies, prominent lower lip, large testes, and obesity.
X-linked intellectual disability, Shashi type is characterised by moderate intellectual deficit, obesity, macroorchidism and a characteristic facies (large ears, a prominent lower lip and puffy eyelids). It has been described in nine boys from two families. Transmission is X-linked and the causative gene has been localised to the q21.3-q27 region of the X chromosome.
Differential diagnosis Differential diagnoses include other primary immunodeficiencies, including 22q11 microdeletion syndrome, HLA class II deficiency and ataxia telangiectasia (see these terms).
Infant respiratory distress syndrome Other names Neonatal respiratory distress syndrome [1] Chest X-ray of a case of IRDS, with fine granular opacities, air bronchograms and bell-shaped thorax Specialty Pediatrics , obstetrics Infantile respiratory distress syndrome ( IRDS ), also called respiratory distress syndrome of newborn , or increasingly surfactant deficiency disorder ( SDD ), [2] and previously called hyaline membrane disease ( HMD ), is a syndrome in premature infants caused by developmental insufficiency of pulmonary surfactant production and structural immaturity in the lungs . ... PMID 23347658 . ^ "Lung Problems in the Premature Baby" . 2012-03-15. ^ "Infant Respiratory Distress Syndrome. IRDS information" . ^ Snyder JM, Mendelson CR (1987). ... "Surfactant therapy and nasal continuous positive airway pressure for newborns with respiratory distress syndrome. Danish-Swedish Multicenter Study Group". ... "Nasal continuous positive airway pressure and early surfactant therapy for respiratory distress syndrome in newborns of less than 30 weeks' gestation" . ... "Prediction of respiratory distress syndrome by the microbubble stability test on gastric aspirates in newborns of less than 32 weeks' gestation".
The A249E mutation had previously been identified in a family with Klippel-Feil syndrome (118100), in 1 proband with isolated microphthalmia (MCOP4; 613094), and in a patient with coloboma and postaxial polydactyly without vertebral defects.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-12 (LCA12) is caused by homozygous mutation in the RD3 gene (180040) on chromosome 1q32. For a general phenotypic description and a discussion of genetic heterogeneity of LCA, see LCA1 (204000). Clinical Features Friedman et al. (2006) identified a sister and brother with Leber congenital amaurosis from a consanguineous Indian family. Both probands had had poor vision since birth. Nystagmus and atrophic lesions in the macular area with pigment migration were found on examination. Preising et al. (2012) reported a large consanguineous Kurdish family with LCA, in which 6 of 7 affected individuals were available for study.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-9 (LCA9) is caused by homozygous or compound heterozygous mutation in the NMNAT1 gene (608700) on chromosome 1p36. For a general discussion of the phenotypic and genetic heterogeneity in Leber congenital amaurosis, see LCA1 (204000). Description Early-onset neurodegeneration in the human retina can lead to Leber congenital amaurosis (LCA), the most severe human form of inherited photoreceptor-neuron degeneration resulting in congenital blindness, with an incidence of approximately 1 in 80,000 (summary by Koenekoop et al., 2012). NMNAT1 (608700) mutations consistently cause severe and rapidly progressive macular degeneration leading to severe central atrophy with an appearance of congenital macular coloboma in the neonatal period, as well as an unusual early-onset atrophy of the optic nerve (Perrault et al., 2012). Clinical Features Koenekoop et al. (2012) reexamined affected individuals from 8 families with Leber congenital amaurosis in whom they had identified mutations in the NMNAT1 gene (608700), which is ubiquitously expressed (see MOLECULAR GENETICS).
A number sign (#) is used with this entry because Leber congenital amaurosis-5 (LCA5) is caused by homozygous mutation in the gene encoding lebercilin (LCA5; 611408) on chromosome 6q14. For a general phenotypic description and a discussion of genetic heterogeneity of LCA, see LCA1 (204000). Clinical Features Dharmaraj et al. (2000) described a multigenerational kindred of Old Order River Brethren, a religious isolate descended from Swiss immigrants to America in the 1750s (Brechbill, 1972), segregating Leber congenital amaurosis. LCA in this kindred was not associated with multisystem abnormalities. Renal function remained normal. Neurologic and hepatic function were within normal limits.
Because LCA manifests very early in life and results in profound vision loss, patients with mutations in other syndromic or nonsyndromic eye disease genes may receive an initial diagnosis of LCA, prior to development of syndromic features or before more thorough phenotyping can be performed (see, e.g., Senior-Loken syndrome-5, 609254). ... In a family reported by Rahn et al. (1968), there were cigarette-paper scars and stretchable skin suggesting Ehlers-Danlos syndrome. Hayasaka et al. (1986) found hyperthreoninemia, hyperthreoninuria, hepatomegaly, and mental and physical retardation in a brother and sister with Leber congenital amaurosis. ... Ehara et al. (1997) reported a previously undescribed autosomal recessive syndrome in 4 Japanese children from 2 unrelated families.
Leber congenital amaurosis (LCA) is an eye disorder that primarily affects the retina . People with this condition typically have severe visual impairment beginning in infancy. Other features include photophobia , involuntary movements of the eyes (nystagmus), and extreme farsightedness. The pupils also do not react normally to light. Additionally, the cornea may be cone-shaped and abnormally thin ( keratoconus ). Franceschetti's oculo-digital sign is characteristic of Leber congenital amaurosis.
A number sign (#) is used with this entry because Leber congenital amaurosis-11 (LCA11) is caused by heterozygous mutation in the IMPDH1 gene (146690) on chromosome 7q32. Heterozygous mutation in the IMPDH1 gene can also cause retinitis pigmentosa-10 (RP10; 180105). Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009).
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-16 (LCA16) is caused by homozygous mutation in the KCNJ13 gene (603208) on chromosome 2q37. For a general phenotypic description and a discussion of genetic heterogeneity of Leber congenital amaurosis, see LCA1 (204000). Clinical Features Sergouniotis et al. (2011) studied a consanguineous Middle Eastern family in which 2 brothers had nystagmus at birth and were diagnosed with Leber congenital amaurosis shortly thereafter. Poor night vision and difficulty reading print from an early age was reported for both patients; gradual progression of visual problems affecting central and peripheral vision was also noted. Both patients had bilateral cataract surgery in their third decade. Funduscopy revealed significant pigment in the retinal pigment epithelium (RPE), in a configuration unlike that of typical retinitis pigmentosa (see 268000).
A number sign (#) is used with this entry because Leber congenital amaurosis-7 can be caused by heterozygous or homozygous mutation in the CRX gene (602225) on chromosome 19q13. Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009). For a general description and a discussion of genetic heterogeneity of LCA, see 204000.
A number sign (#) is used with this entry because Leber congenital amaurosis-15 and juvenile retinitis pigmentosa are caused by homozygous or compound heterozygous mutation in the TULP1 gene (602280) on chromosome 6p21.3. Description Autosomal recessive childhood-onset severe retinal dystrophy is a heterogeneous group of disorders affecting rod and cone photoreceptors simultaneously. The most severe cases are termed Leber congenital amaurosis, whereas the less aggressive forms are usually considered juvenile retinitis pigmentosa (summary by Gu et al., 1997). Mutation in TULP1 can also cause a form of autosomal recessive retinitis pigmentosa (RP14; 600132). For a general phenotypic description and a discussion of the genetic heterogeneity of Leber congenital amaurosis, see LCA1 (204000); for retinitis pigmentosa, see 268000.
A number sign (#) is used with this entry because Leber congenital amaurosis-8 (LCA8) is caused by homozygous or compound heterozygous mutation in the CRB1 gene (604210) on chromosome 1q31. Homozygous or compound heterozygous mutation in CRB1 can also cause retinitis pigmentosa-12 (RP12; 600105). Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009).
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-6 (LCA6) is caused by homozygous or compound heterozygous mutation in the RPGRIP1 gene (605446) on chromosome 14q11. Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009). For a general description and a discussion of genetic heterogeneity of LCA, see 204000.
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-2 (LCA2) is caused by homozygous or compound heterozygous mutation in the RPE65 gene (180069) on chromosome 1p31. Mutations in this gene also cause retinitis pigmentosa (RP20; 613794). Description Leber congenital amaurosis comprises a group of early-onset childhood retinal dystrophies characterized by vision loss, nystagmus, and severe retinal dysfunction. Patients usually present at birth with profound vision loss and pendular nystagmus. Electroretinogram (ERG) responses are usually nonrecordable. Other clinical findings may include high hypermetropia, photodysphoria, oculodigital sign, keratoconus, cataracts, and a variable appearance to the fundus (summary by Chung and Traboulsi, 2009).
LCA may be associated with mutations in genes linked to syndromes presenting with neurodevelopmental delay, intellectual disability, oculomotor apraxia-type behavior (difficulty moving the eye) and renal dysfunction. ... Differential diagnosis Differential diagnosis includes retinitis pigmentosa, Alström syndrome, Joubert syndrome, Stargardt disease, Senior-Loken syndrome, Conorenal syndrome and infantile neuronal ceroid lipofuscinosis.
Detailed CT scanning revealed no molar-tooth sign, no cerebellar atrophy, and no structural signs of Joubert syndrome (see 213300). McEwen et al. (2007) found that affected individuals from the family reported by den Hollander et al. (2006) had severely impaired olfactory function, whereas heterozygous mutation carriers had mild to severe microsomia. ... Mapping Using linkage analysis, den Hollander et al. (2006) assigned the gene responsible for LCA in a consanguineous French Canadian family with 4 affected sibs to chromosome 12q21-q22, in a region containing 15 genes, including CEP290 (610142). Joubert syndrome-5 (610188), which is due to mutations in the CEP290 gene, is associated in all patients with congenital amaurosis or retinitis pigmentosa. ... The patients had no neurologic symptoms typical of Joubert syndrome, had normal cognitive function, and showed no clinical signs of renal disease. ... The results suggested a complete loss of function of both CEP290 alleles leads to Joubert syndrome, whereas patients with LCA10 have a small amount of residual CEP290 activity.
Sorsby and Williams (1960) observed a family with multiple cases of retinal aplasia in which inheritance was autosomal dominant. 'Retinal aplasia' is the British term for what is called 'congenital amaurosis' on the continent. Much genetic heterogeneity exists as evidenced by the demonstration of both autosomal dominant and autosomal recessive forms. This disorder, which might be called an autosomal dominant form of Leber amaurosis congenita, must be very rare. Heckenlively (1988) reported a 6-generation family with a severe progressive retinal degeneration beginning in infancy.
A number sign (#) is used with this entry because of evidence that this form of common variable immunodeficiency, referred to here as CVID6, is caused by homozygous mutation in the CD81 gene (186845) on chromosome 11p. One such patient has been reported. For a general description and a discussion of genetic heterogeneity of common variable immunodeficiency, see CVID1 (607594). Clinical Features Van Zelm et al. (2010) reported a 6-year-old Moroccan girl, born of consanguineous parents, who had recurrent respiratory tract infections in the first 2 years of life. At age 3.5 years, she developed an acute glomerulonephritis with proteinuria, a purpuric rash, and arthralgias, associated with deposition of IgA and C3 (120700) and resulting in renal failure. She also had hepatomegaly and thrombocytopenia associated with antiplatelet antibodies.
Overview Common variable immunodeficiency (CVID) is an immune system disorder that causes you to have low levels of the proteins that help fight infections. If you have CVID , you'll likely have repeated infections in your ears, sinuses and respiratory system. You'll also have an increased risk of digestive disorders, autoimmune disorders, blood disorders and cancer. CVID can be inherited, or you can develop it during your lifetime. Symptoms The severity of symptoms can vary greatly between people with CVID . Symptoms of common variable immunodeficiency may appear during childhood or adolescence, though many people don't experience them until adulthood.
A number sign (#) is used with this entry because of evidence that autosomal dominant common variable immunodeficiency-12 (CVID12) is caused by heterozygous mutation in the NFKB1 gene (164011) on chromosome 4q24. Description Common variable immunodeficiency-12 is an autosomal dominant primary immunodeficiency characterized by recurrent infections, mainly respiratory, associated with hypogammaglobulinemia. The disorder shows a highly variable age at onset and highly variable disease severity, even within the same family. Some patients have features of autoimmunity (summary by Fliegauf et al., 2015). For a general description and a discussion of genetic heterogeneity of common variable immunodeficiency, see CVID1 (607594).
A number sign (#) is used with this entry because common variable immunodeficiency-10 (CVID10) is caused by heterozygous mutation in the NFKB2 gene (164012) on chromosome 10q24. Description Common variable immunodeficiency-10 is an autosomal dominant primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency (summary by Chen et al., 2013). For a general description and a discussion of genetic heterogeneity of common variable immunodeficiency, see CVID1 (607594).
Common variable immunodeficiency (CVID) is a group of disorders characterized by low levels of a type of protein known as immunoglobulins (Ig). Because of low level of Ig, the immune system cannot make antibodies that fight bacteria, viruses or other toxins in the body. This leads to frequent infections, particularly in the sinuses, lungs, and digestive tract. Symptoms most commonly begin in early adulthood but can occur at any age. While in most cases the cause of CVID is unknown, a genetic change has been found in about one-third of cases.
A number sign (#) is used with this entry because this form of common variable immunodeficiency (CVID), referred to here as CVID5, is caused by homozygous mutation in the CD20 gene (MS4A1; 112210) on chromosome 11q13. For a general description and a discussion of genetic heterogeneity of common variable immunodeficiency, see CVID1 (607594). Clinical Features Kuijpers et al. (2010) reported a Turkish girl, born of consanguineous parents, who developed recurrent respiratory infections and bronchopneumonia at age 2 years. At onset, she had low IgG and low IgA levels. During a follow-up of 4 years, she showed normal IgM and IgA levels, but low IgG levels. Laboratory studies showed normal numbers of B cells, but persistent hypogammaglobulinemia, reduced circulating memory B cells, and complete lack of surface CD20 on B cells.
A number sign (#) is used with this entry because this form of common variable immunodeficiency (CVID), referred to here as CVID2, is caused by heterozygous, homozygous, or compound heterozygous mutation in the TNFRSF13B gene (604907), which encodes the transmembrane activator and CAML interactor (TACI), on chromosome 17p11.2. Selective IgA deficiency-2 (IGAD2; 609529) can also be caused by mutation in the TNFRSF13B gene. For a general description and a discussion of genetic heterogeneity of common variable immunodeficiency, see CVID1 (607594). Clinical Features Salzer et al. (2005) reported 2 unrelated families with a diagnosis of common variable immunodeficiency. All affected individuals had hypogammaglobulinemia with low serum IgG, IgM, and IgA, and recurrent infections, including otitis media, respiratory tract infections, and gastrointestinal tract infections.
Common variable immunodeficiency (CVID) comprises a heterogeneous group of diseases characterized by a significant hypogammaglobulinemia of unknown cause, failure to produce specific antibodies after immunizations and susceptibility to bacterial infections, predominantly caused by encapsulated bacteria. Epidemiology Prevalence is estimated at 1/25,000 among Caucasians and CVID affects men and women equally. Clinical description While some patients are diagnosed with CVID in early childhood, the major peak of onset lies between the second and third decade of life, frequently with several years delay between onset and diagnosis. Over 98% of patients present with recurrent bronchitis, sinusitis, otitis and pneumonia, and chronic pulmonary damage is the major complication. About 25% of patients develop autoimmune phenomena; immune thrombocytopenic purpura (ITP) and autoimmune hemolytic anemia (AIHA) are the most common (see these terms).
Rosen and Bougas (1963) reported the case of a woman with recurrent infection, marked elevation of 19S gamma globulin, and virtual absence of 7S. Feldman et al. (1975) found that 12 relatives had a variable immunodeficiency. Ten had elevation of IgM, combined with deficiency of IgG and IgA in 3 and deficiency of one or the other in 2. Five had only elevated IgM and 2 had normal IgM but deficiency of IgG and IgA. No male-to-male transmission was observed. Immunology - Variable immunodeficiency - Increased IgM - IgG deficiency - IgA deficiency Misc - Recurrent infection Inheritance - Autosomal dominant ▲ Close
The disorder was consistent with a clinical diagnosis of common variable immunodeficiency syndrome. One family included 3 Colombian sibs examined at 35, 33, and 49 years of age, respectively.
A number sign (#) is used with this entry because this form of common variable immunodeficiency (CVID), referred to here as CVID1, is caused by homozygous mutation in the ICOS gene (604558) on chromosome 2q33. Description Common variable immunodeficiency (CVID) is a clinically and genetically heterogeneous group of disorders characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections, and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B cells are usually in the normal range, but can be low. Most individuals with CVID have onset of infections after age 10 years. CVID represents the most common form of primary immunodeficiency disorders and is the most common form of primary antibody deficiency.
At age 57, he received a clinical diagnosis of common variable immunodeficiency syndrome after recurrent pneumonia. His 80-year-old sister had an unremarkable medical history except for a severe Herpes zoster infection at age 70 and recent recurrent pneumonia.
Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( February 2017 ) Congenital bilateral perisylvian syndrome Other names CBPS This condition is inherited in an X-linked dominant manner. [1] Specialty Neurology Congenital bilateral perisylvian syndrome ( CBPS ) is a rare neurological disease characterized by paralysis of certain facial muscles and epileptic seizures . ... These include pachygyria , double cortex syndrome , and lissencephaly , all of which are classified along with CBPS as neuronal migration disorders . [2] Diagnostic tests for CBPS include electroencephalograms , CT scanning , and magnetic resonance imaging . [2] Treatment [ edit ] CBPS is commonly treated with anticonvulsant therapy to reduce seizures. ... Retrieved 19 August 2017 . ^ a b c d e f g h "Congenital Bilateral Perisylvian Syndrome – NORD (National Organization for Rare Disorders)" .
A number sign (#) is used with this entry because bilateral perisylvian polymicrogyria with autosomal recessive inheritance is caused by homozygous deletion of one 15-bp tandem repeat in a regulatory region of exon 1m of the ADGRG1 gene (604110) on chromosome 16q21. Mutations in the ADGRG1 gene also cause bilateral frontoparietal polymicrogyria (606854). Description Autosomal recessive bilateral perisylvian polymicrogyria is characterized by strikingly restricted polymicrogyria limited to the cortex surrounding the Sylvian fissure. Affected individuals have intellectual and language difficulty and seizures, but no motor disability (Bae et al., 2014). Clinical Features Bae et al. (2014) examined more than 1,000 individuals with gyral abnormalities and identified 5 individuals from 3 families (1 Turkish and 2 Irish American) with strikingly restricted polymicrogyria limited to the cortex surrounding the Sylvian fissure, which suggested a rare, genetically distinctive condition.
It presents with developmental delay, intellectual disability, seizures and various neurological impairments and may be isolated or comprise a clinical feature of many genetic syndromes. It may also be associated with perinatal cytomegalovirus infection.
Genetic causes may include mutations in single genes and contiguous gene disorders such as 22q11.2 deletion syndrome . BPP has also been reported in association with twin pregnancy complications. ... The quality of life and life expectancy for people with BPP are not well-described but may depend on severity of symptoms, whether complications develop, and whether other birth defects or an underlying syndrome are present.
A number sign (#) is used with this entry because of evidence that perisylvian polymicrogyria, cerebellar hypoplasia, and arthrogryposis (PMGYCHA) is caused by compound heterozygous mutation in the PI4KA gene (600286) on chromosome 22q11. One such family has been reported. Clinical Features Pagnamenta et al. (2015) reported a family in which 3 fetuses were all diagnosed in utero with multiple congenital anomalies resulting in the termination of pregnancy between 16 and 34 weeks' gestation. All fetuses had bilateral perisylvian polymicrogyria and cerebellar hypoplasia or dysplasia. One had a small pons. Additional features included severe talipes equinovarus, externally rotated hips, and variable contractures of the limbs or fingers. Three had micrognathia and 2 had dolichocephaly. Muscle histology was reported to be within normal limits.
Bilateral perisylvian PMG (BPP) often results in a typical clinical syndrome that is manifested by mild mental retardation, epilepsy, and pseudobulbar palsy, which causes difficulties with expressive speech and feeding (Kuzniecky et al., 1993). ... Clinical Features Kuzniecky et al. (1993) studied 31 patients with a congenital neurologic syndrome characterized by pseudobulbar palsy, cognitive deficits, and bilateral perisylvian abnormalities on imaging studies. ... The clinical manifestations of pseudobulbar palsy, congenital facio-pharyngo-glosso-masticatory diplegia represented developmental Foix-Chavany-Marie syndrome, or bilateral anterior opercular syndrome, resulting from bilateral opercular lesions. ... Clark and Neville (2008) noted that there is significant phenotypic overlap between PMG and Worster-Drought syndrome (185480) and that the disorders may be part of a phenotypic spectrum. ... In addition, a severe and unlayered form of PMG is a prominent feature of Zellweger syndrome (see 214100). Inheritance Familial recurrence of PMG in a pattern consistent with X-linked inheritance was reported by Borgatti et al. (1999) and Guerreiro et al. (2000).
Neurogenic thoracic outlet syndrome (NTOS) is a form of thoracic outlet syndrome (TOS; see this term) that presents with pain, paresthesias and weakness in an upper extremity and is divided into true NTOS and disputed NTOS. ... Differential diagnosis Differential diagnoses include arterial and venous TOS (see these terms), cervical radiculopathy, carpal tunnel syndrome or any disorder involving nerve fibers derived from C8 or T1 nerve roots such as cubital tunnel syndrome.
Hypertelorism with ocular anomalies are also observed, generally with normal neuropsychological development. Epidemiology Pai syndrome (PS) has been reported in 67 patients to date, however, the incidence seems to be underestimated. ... Differential diagnosis Differential diagnoses include Loeys-Dietz syndrome, oculocerebrocutaneous syndrome, frontonasal dysplasia, Goldenhar syndrome, as well as a variety of chromosomal anomalies.
Description Pai syndrome is characterized by mild hypertelorism, midline cleft lip, nasal and facial polyps, pericallosal lipoma, ocular anomalies, and normal neuropsychologic development (Guion-Almeida et al., 2007). Clinical Features Pai syndrome is a rare disorder combining median cleft upper lip and polyps of the facial skin and nasal mucosa (Sharma, 1974; Ponniah, 1977; Nakamura et al., 1985; Pai et al., 1987). In addition, midline lipomas of the central nervous system are part of the syndrome, as described by Pai et al. (1987). ... Al-Mazrou et al. (2001) described Pai syndrome in one of monozygotic female twins. ... Guion-Almeida et al. (2007) reported 7 South American patients with Pai syndrome. The phenotype was clinically variable and 5 of the patients were severely affected.
A number sign (#) is used with this entry because of evidence that immunodeficiency-centromeric instability-facial anomalies syndrome-4 (ICF4) is caused by homozygous or compound heterozygous mutation in the HELLS gene (603946) on chromosome 10q23. Description Immunodeficiency-centromeric instability-facial anomalies syndrome-4 is an autosomal recessive disorder characterized by recurrent infections in childhood and variable dysmorphic facial features. ... For a discussion of genetic heterogeneity of immunodeficiency-centromeric instability-facial anomalies syndrome, see ICF1 (242860). Clinical Features Thijssen et al. (2015) reported 5 patients from 4 unrelated families with an immunodeficiency syndrome characterized by recurrent respiratory infections and associated with hypo- or agammaglobulinemia with normal B cells.