2p21 microdeletion syndrome without cystinuria is a rare partial autosomal monosomy characterized by weak fetal movements, severe infantile hypotonia and feeding difficulties that spontaneously improve with time, urogenital abnormalities (hypospadias or hypoplastic labia majora), global development delay, mild intellectual disability and facial dysmorphism (dolichocephaly, frontal bossing, bilateral ptosis, midface retrusion, open mouth with tented upper lip vermilion).
A rare, genetic, syndromic intellectual disability disorder characterized by variable degrees of intellectual disability, behavioral problems (including attention deficit and hyperactivity disorder, autism spectrum disorder, and aggressiveness), an altered sleeping pattern, and delayed speech and language development associated with disruption of ankryin-3 ( ANK3 gene).
A number sign (#) is used with this entry because of evidence that autosomal recessive mental retardation-37 (MRT37) is caused by homozygous mutation in the ANK3 gene (600465) on chromosome 10q21. One such family has been reported. Clinical Features Iqbal et al. (2013) reported 3 sibs, born of consanguineous Pakistani parents, with moderate intellectual disability (IQ less than 50). The patients were 25, 22, and 18 years of age at the time of the report. All had delayed global development with speech delay, hypotonia, spasticity, and a sleep disorder. All had severe behavioral abnormalities, including aggression, hyperactivity, and grinding of the teeth.
A rare X-linked syndromic intellectual disability characterized by congenital sensorineural hearing loss, varying degrees of intellectual disability, short stature, and dysmorphic facial features (such as telecanthus, epicanthic folds, broad nasal root, malar hypoplasia, low-set ears, dental anomalies, and micrognathia).
Mapping Martin et al. (2000) determined that the 3 males with the deafness-mental retardation syndrome described by them shared a haplotype between DXS1003 and DXS1220, a 68-Mb interval spanning Xq1-q21. ... Molecular Genetics Associations Pending Confirmation For discussion of a possible association between Martin-Probst X-linked mental retardation syndrome and variation in the RAB40AL gene, see 300405.0001.
Description Imerslund-Grasbeck syndrome is a form of congenital megaloblastic anemia due to vitamin B12 deficiency caused by a defect in the vitamin B12/intrinsic factor receptor. ... Imerslund and Bjornstad (1963) and Lamy et al. (1961) reported on the syndrome of chronic relapsing megaloblastic anemia and permanent proteinuria. ... Persistent proteinuria appears to be an integral part of the syndrome (Mohamed et al., 1966). The latter authors gave a genetic analysis of published cases. ... Spurling et al. (1964) described 2 Baltimore sisters with this syndrome who had proteinuria. Their parents were fourth cousins. ... Cytogenetics Celep et al. (1996) described Imerslund-Grasbeck syndrome in 3 Turkish sibs with first-cousin parents.
Imerslund-Grasbeck syndrome (IGS) or selective vitamin B12 (cobalamin) malabsorption with proteinuria is a rare autosomal recessive disorder characterized by vitamin B12 deficiency commonly resulting in megaloblastic anemia, which is responsive to parenteral vitamin B12 therapy and appears in childhood. Epidemiology The syndrome was first described in Finland and Norway where the prevalence is about 1/200 000.
Imerslund–Gräsbeck syndrome Other names Imerslund–Najman–Gräsbeck syndrome, Imerslund–Gräsbeck disease (IGS or INGS), Imerslund syndrome, Congenital cobalamin malabsorption or Autosomal recessive megaloblastic anemia (MGA1) This condition is inherited via an autosomal recessive manner Imerslund–Gräsbeck syndrome , is a rare autosomal recessive , familial form of vitamin B12 deficiency caused by malfunction of the " Cubam " receptor located in the terminal ileum. ... A defect in either of these protein components can cause this syndrome. This is a rare disease, with a prevalence about 1 in 200,000, [1] and is usually seen in patients of European ancestry. ... To understand the basic pathophysiology of Imerslund–Gräsbeck syndrome, it is imperative to understand the absorption of vitamin B 12 . ... "Imerslund-Gräsbeck syndrome (selective vitamin B 12 malabsorption with proteinuria)" . ... Blood, 103(5), 1573-1579. ^ Grasbeck, R. (2006). Imerslund-Grasbeck syndrome (selective vitamin B12 malabsorption with proteinuria).
Osteopetrosis-hypogammaglobulinemia syndrome is an extremely rare primary bone dysplasia with increased bone density disorder characterized by severe osteoclast-poor osteopetrosis associated with hypogammaglobulinemia.
A number sign (#) is used with this entry because of evidence that autosomal recessive osteopetrosis-7 (OPTB7) is caused by homozygous or compound heterozygous mutation in the TNFRSF11A gene (603499) on chromosome 18q21. For a general phenotypic description and a discussion of genetic heterogeneity of autosomal recessive osteopetrosis, see OPTB1 (259700). Clinical Features Guerrini et al. (2008) described 8 patients from 7 unrelated families with severe osteoclast-poor osteopetrosis, 1 of whom (patient 2) was an Argentinian male infant who had previously been studied by Blair et al. (2004) (subject 2). Serum immunoglobulin levels were found to be reduced in 3 of 4 patients who underwent assessment; 2 of the 3 hypogammaglobulinemic patients failed to produce antibodies after a full course of tetanus toxoid vaccination. Bone biopsy specimens from 3 patients showed extensive trabecular structures, with retention of large areas of cartilage, complete absence of multinucleated cells, and lack of osteoclastic resorption; in 1 patient, the possible presence of small mononuclear osteoclasts was excluded by histochemical staining for tartrate-resistant acid phosphatase (TRAP).
Osteopetrosis refers to a group of rare, inherited skeletal disorders characterized by increased bone density and abnormal bone growth. Symptoms and severity can vary greatly, ranging from neonatal onset with life-threatening complications (such as bone marrow failure) to the incidental finding of osteopetrosis on X-ray. Depending on severity and age of onset, features may include fractures, short stature, compressive neuropathies (pressure on the nerves), hypocalcemia with attendant tetanic seizures, and life-threatening pancytopenia . In rare cases, there may be neurological impairment or involvement of other body systems. Osteopetrosis may be caused by mutations in at least 10 genes. Inheritance can be autosomal recessive, autosomal dominant, or X-linked recessive with the most severe forms being autosomal recessive.
Lipodystrophy-intellectual disability-deafness syndrome is an extremely rare form of genetic lipodystrophy (see this term), reported in 3 patients from one family to date, characterized by generalized congenital lipodystrophy, low birth weight, progressive sensorineural deafness occurring in childhood, intellectual deficit, progressive osteopenia, delayed skeletal maturation, skeletal abnormalities described as slender, undermineralized tubular bones, and dense metaphyseal striations in the distal femur, ulna and radius of older patients.
Clinical Features Rajab et al. (2003) reported 3 patients, including 2 sibs, with congenital generalized lipodystrophy, sensorineural deafness, low birth weight, short stature, delayed cognitive development, and progressive bone changes characterized by overtubulation and rarefaction of long bones with dense metaphyseal striations occurring in adolescence. No abnormalities of lipid or carbohydrate metabolism, hepatosplenomegaly, acanthosis nigricans, or hirsutism were found. Inheritance Rajab et al. (2003) suggested that the occurrence of this disorder in sibs born to consanguineous parents and the observation of a third patient from the same Omani tribal unit suggested autosomal recessive inheritance. INHERITANCE - Autosomal recessive GROWTH Height - Short stature Weight - Low birth weight - Thin body habitus Other - Failure to thrive - Intrauterine growth retardation HEAD & NECK Face - Progeroid facial appearance (onset childhood) - Maxillary hypoplasia Ears - Sensorineural hearing loss (onset early childhood) Eyes - Deep-set eyes CHEST External Features - Long thorax SKELETAL - Progressive osteopenia - Delayed bone age Pelvis - Short femoral necks Limbs - Thin limbs with prominent joints - Cubitus valgus - Genua valgum - Slender long bones with narrow diaphyses - Dense longitudinal metaphyseal striations (distal femur, radius, and ulna) Hands - Disharmonious carpal bone SKIN, NAILS, & HAIR Skin - Sparse axillary and facial hair MUSCLE, SOFT TISSUES - Congenital generalized lipodystrophy NEUROLOGIC Central Nervous System - Mental retardation - Seizures ▲ Close
Clinical Features Somech et al. (2008) described 2 sisters, born of first-cousin parents of Sri Lankan descent, who presented in infancy with immunodeficiency, gonadal dysgenesis, and fatal pulmonary fibrosis. The infants displayed no dysmorphic features. Immune studies demonstrated combined humoral and cellular abnormalities including reduced immunoglobulin production, absence of lymphoid tissue, markedly reduced T lymphocyte numbers and function, and reduced newly thymus-derived T cells. Both infants succumbed to rapidly progressive pulmonary fibrosis; autopsy revealed streak ovaries bearing no discernible oocytes. Molecular Genetics In 2 sisters with normal 46,XX karyotype and immunodeficiency, gonadal dysgenesis, and fatal pulmonary fibrosis, Somech et al. (2008) performed FISH analysis that showed no deletion or duplication for the X centromeric region, the SRY gene (480000), and chromosome 22q11.2. Analysis of genes known to be associated with severe immune defects in infancy (IL7R, 146661; JAK3, 600173; CD3, see 186740) and ovarian dysgenesis (FSHR; 136435) showed no abnormality.
Cerebellar ataxia - areflexia - pes cavus - optic atrophy - sensorineural hearing loss (CAPOS syndrome) is a rare autosomal dominant neurological disorder characterized by early onset cerebellar ataxia, associated with areflexia, progressive optic atrophy, sensorineural deafness, a pes cavus deformity, and abnormal eye movements.
Epidemiology Autosomal dominant optic atrophy plus syndrome (ADOA plus) accounts for approximately 20% of all ADOA cases. ... Differential diagnosis Differential diagnosis includes several other syndromic hereditary optic neuropathies that may have bilateral manifestations associated with extra ocular features and that presents with a similar phenotype, such as Autosomal dominant Charcot-Marie-Tooth disease type 2A, Leber hereditary optic neuropathy, Wolfram syndrome and Wolfram-like syndrome.
Description Optic atrophy-8 (OPA8) is an autosomal dominant neurologic disorder characterized by progressive visual loss during the first or second decade of life. Some patients may have additional features, mainly late-onset sensorineural hearing loss. For a discussion of genetic heterogeneity of optic atrophy, see OPA1 (165500). Clinical Features Carelli et al. (2011) reported a large multigenerational kindred from the Italian region of Emilia-Romagna in which 53 individuals had with optic neuropathy. Five affected adults were studied in detail. Onset of visual loss occurred between 6 and 21 years of age, and symptoms included central scotoma, diffuse reduction in the retinal nerve fiber layer, and abnormal visual evoked potentials.
Hagemoser et al. (1989) reported 2 unrelated families with a disorder characterized by optic atrophy, hearing loss, and peripheral neuropathy. In the first family, there were 13 affected members spanning 4 generations with an instance of male-to-male transmission. Most patients had onset of bilateral hearing loss and visual loss with optic atrophy by school-age. Onset of neurologic features occurred only in a subset of patients as adults, and consisted primarily of decreased vibratory sensation and hyporeflexia in the lower limbs. Nerve conduction velocities suggested an axonal sensory and motor neuropathy.
Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) syndrome is a neurological disorder. The syndrome gets its name from the different parts of the body it usually affects: Cerebellar ataxia : inflammation of the cerebellum, the part of the brain that helps control the coordination of muscle movement Areflexia: loss of reflexes Pes cavus : high arch in foot Optic atrophy : damage to the optic nerve of the eye Sensorinural hearing loss : damage to the nerves involved in hearing CAPOS syndrome typically begins after a fever-related illness with a sudden episode of ataxia, such as having a hard time walking or coordinating leg or arm movements. ... Pregnancy and delivery may also trigger episodes. Most people with CAPOS syndrome have one to three episodes during their lifetime. ... Though many of the signs and symptoms of CAPOS syndrome get better as the fever and illness improve, some symptoms, including movement problems, may continue.
A number sign (#) is used with this entry because cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) is caused by heterozygous mutation in the ATP1A3 gene (182350) on chromosome 19q13. Heterozygous mutation in the ATP1A3 gene can also cause 2 other neurologic disorders that share some clinical features: dystonia-12 (DYT12; 128235) and alternating hemiplegia of childhood-2 (AHC2; 614820). Description Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) is an autosomal dominant neurologic disorder characterized by early-childhood onset of recurrent episodes of acute ataxic encephalopathy associated with febrile illnesses. These acute episodes tend to decrease with time, but the neurologic sequelae are permanent and progressive, resulting in gait and limb ataxia and areflexia. Affected individuals also develop progressive visual impairment due to optic atrophy and sensorineural hearing loss beginning in childhood.
Fraley syndrome Fraley syndrome Fraley syndrome is a condition where the superior infundibulum of the upper calyx of the kidney is obstructed by the crossing renal (upper or middle section) artery branch, causing distension and dilatation of the calyx and presenting clinically as haematuria and nephralgia (ipsilateral flank pain). [1] [2] [3] [4] Furthermore, when the renal artery obstructs the proximal collecting system, filling defects can occur anywhere in the calyces, pelvis, or ureter. ... ] ^ Zuckier LS, Patel YD, Fine EJ, Koenigsberg M (1988). "Fraley's syndrome: case report and update on current diagnostic methods". ... Retrieved 2019-12-13 . ^ a b c d ePainAssist, Team (2014-07-24). "Fraley's Syndrome: Treatment, Causes, Symptoms, Diagnosis" . ePainAssist . ... J.; Koenigsberg, M. (1988). "Fraley's syndrome: case report and update on current diagnostic methods". ... "Experience in the surgical treatment of Fraley's syndrome". European Urology . 38 (4): 410–414. doi : 10.1159/000020316 .
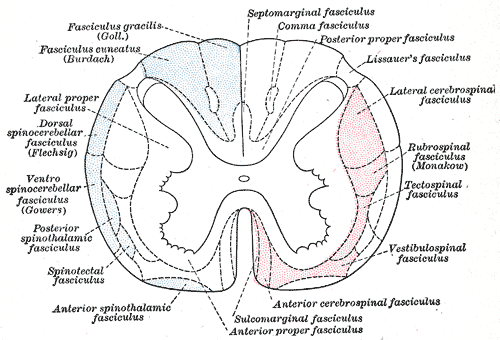
Specialty Neurology Subacute combined degeneration of spinal cord , also known as Lichtheim 's disease [1] [2] or Putnam-Dana syndrome, [3] refers to degeneration of the posterior and lateral columns of the spinal cord as a result of vitamin B 12 deficiency (most common), vitamin E deficiency , [4] and copper deficiency . [5] It is usually associated with pernicious anemia . ... This may be due to a dietary deficiency, malabsorption in the terminal ileum , lack of intrinsic factor secreted from gastric parietal cells, or low gastric pH inhibiting attachment of intrinsic factor to ileal receptors. [9] Vitamin E deficiency , which is associated with malabsorption disorders such as cystic fibrosis and Bassen-Kornzweig syndrome , [10] can cause a similar presentation due to the degeneration of the dorsal columns . [4] Diagnosis [ edit ] Serum vitamin B 12 , methylmalonic acid , Schilling test , and a complete blood count, looking for megaloblastic anemia if there is also folic acid deficiency or macrocytic anemia . ... "Subacute Combined Degeneration of the Cord: Putnam-Dana Syndrome" . European Neurology . 60 . doi : 10.1159/000131715 . ^ a b Agamanolis, Dimitri P. ... Deficiency of the lipid-soluble vitamin E occurs in cases of intestinal malabsorption such as cystic fibrosis, congenital biliary atresia, intestinal resection, and abetalipoproteinemia (Bassen-Kornzweig syndrome). ^ Carmel, Ralph (2007). "The Disappearance of Cobalamin Absorption Testing: A Critical Diagnostic Loss" . ... External links [ edit ] Classification D ICD - 10 : G32.0 , E53.8 ICD - 9-CM : 336.2 , 266.2 MeSH : D052879 DiseasesDB : 12591 External resources MedlinePlus : 000723 v t e Malnutrition Protein-energy malnutrition Kwashiorkor Marasmus Catabolysis Vitamin deficiency B vitamins B 1 Beriberi Wernicke–Korsakoff syndrome Wernicke's encephalopathy Korsakoff's syndrome B 2 Riboflavin deficiency B 3 Pellagra B 6 Pyridoxine deficiency B 7 Biotin deficiency B 9 Folate deficiency B 12 Vitamin B 12 deficiency Other A: Vitamin A deficiency Bitot's spots C: Scurvy D: Vitamin D deficiency Rickets Osteomalacia Harrison's groove E: Vitamin E deficiency K: Vitamin K deficiency Mineral deficiency Sodium Potassium Magnesium Calcium Iron Zinc Manganese Copper Iodine Chromium Molybdenum Selenium Keshan disease Growth Delayed milestone Failure to thrive Short stature Idiopathic General Anorexia Weight loss Cachexia Underweight v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis v t e Focal lesions of the spinal cord General Myelopathy Myelitis Spinal cord compression By location Brown-Séquard syndrome Posterior cord syndrome Anterior cord syndrome Central cord syndrome Cauda equina syndrome Other Polio Demyelinating disease Transverse myelitis Tropical spastic paraparesis Epidural abscess Syringomyelia Syringobulbia Morvan's syndrome Sensory ataxia Tabes dorsalis Abadie's sign Subacute combined degeneration of spinal cord Vascular myelopathy Anterior spinal artery syndrome Foix–Alajouanine syndrome
A number sign (#) is used with this entry because of evidence that peeling skin syndrome-5 (PSS5) is caused by homozygous mutation in the SERPINB8 (601697) on chromosome 18q22. Description Peeling skin syndrome-5 (PSS5) is characterized by superficial peeling of the dorsal and palmar skin of the hands and feet; the skin of the forearms and legs may also be involved. ... For a general phenotypic description and a discussion of genetic heterogeneity of peeling skin syndrome, see PSS1 (270300). Clinical Features Pigors et al. (2016) studied 3 unrelated families with exfoliative ichthyosis. ... Mapping By high-resolution homozygosity mapping in affected members of a Tunisian family with peeling skin syndrome, Pigors et al. (2016) identified a shared region of homozygosity on chromosome 18 between SNPs rs1944983 and rs2194631.
A number sign (#) is used with this entry because of evidence that peeling skin syndrome-4 (PSS4) is caused by homozygous mutation in the CSTA gene (184600) on chromosome 3q21. For a general phenotypic description and a discussion of genetic heterogeneity of peeling skin syndrome, see PSS1 (270300). Clinical Features Hatsell et al. (2003) reported 2 related Bedouin families in which 5 individuals had peeling skin of the hands and feet as well as generalized dry scaling skin over the remainder of the body. ... Pavlovic et al. (2012) described a large Jordanian American family in which 10 members had an acral form of peeling skin syndrome. The proband was a 24-year-old man with a lifelong history of peeling skin on his hands and feet that worsened with exposure to heat, friction, perspiration, or water, and was also associated with pruritus. ... In a patient with an acral form of peeling skin syndrome from a large Jordanian American family that was originally reported by Pavlovic et al. (2012) and in which no mutation was found in the TGM5 (603805), CDSN (602593), KRT14 (148066), or KRT5 (148040) genes, Krunic et al. (2013) performed whole-exome sequencing and identified a homozygous nonsense mutation in the CSTA gene (K22X; 184600.0003).
Exfoliative ichthyosis is an inherited, non-syndromic, congenital ichthyosis disorder characterized by the infancy-onset of palmoplantar peeling of the skin (aggravated by exposure to water and by occlusion) associated with dry, scaly skin over most of the body.
It is associated with variants of type VI collagen , it is commonly associated with muscle weakness and respiratory problems, though cardiac issues are not associated with this type of CMD. [4] [5] It is named after Otto Ullrich, who is also known for the Ullrich-Turner syndrome . [6] Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 3.1 Differential diagnosis 4 Treatment 4.1 Prognosis 5 Research 6 See also 7 References 8 Further reading 9 External links Signs and symptoms [ edit ] The presentation of Ullrich congenital muscular dystrophy in an affected individual is as follows: [2] [7] Muscle weakness Difficulty walking Contractures (predominantly in proximal muscles, e.g. neck) Joint looseness (predominantly in distal joints) Genetics [ edit ] In terms of the genetics of Ullrich congenital muscular dystrophy, there are mutations in the genes COL6A1 , COL6A2 , and COL6A3 . ... Alpha 1 subunit of type VI collagen is the encoded protein. [9] Diagnosis [ edit ] Micrograph hyperkeratosis In terms of the diagnosis of Ullrich congenital muscular dystrophy upon inspection follicular hyperkeratosis , may be a dermatological indicator, additionally also serum creatine kinase may be mildly above normal. [5] Other exams/methods to ascertain if the individual has Ullrich congenital muscular dystrophy are: [ medical citation needed ] MRI Biopsy muscle Genetic testing Differential diagnosis [ edit ] This includes [10] Autosomal recessive myosclerosis Bethlem myopathy Ehlers–Danlos syndrome Emery–Dreifuss muscular dystrophy Limb-girdle muscular dystrophy RYR1-associated multiminicore disease Treatment [ edit ] Scoliosis X-ray Treatment for Ullrich congenital muscular dystrophy can consist of physical therapy and regular stretching to prevent and reduce contractures . ... External links [ edit ] Classification D ICD - 10 : G71.2 OMIM : 254090 MeSH : C537521 DiseasesDB : 33679 External resources GeneReviews : Collagen Type VI-Related Disorders Orphanet : 75840 Scholia has a topic profile for Ullrich congenital muscular dystrophy . v t e Diseases of muscle , neuromuscular junction , and neuromuscular disease Neuromuscular- junction disease autoimmune Myasthenia gravis Lambert–Eaton myasthenic syndrome Neuromyotonia Myopathy Muscular dystrophy ( DAPC ) AD Limb-girdle muscular dystrophy 1 Oculopharyngeal Facioscapulohumeral Myotonic Distal (most) AR Calpainopathy Limb-girdle muscular dystrophy 2 Congenital Fukuyama Ullrich Walker–Warburg XR dystrophin Becker's Duchenne Emery–Dreifuss Other structural collagen disease Bethlem myopathy PTP disease X-linked MTM adaptor protein disease BIN1-linked centronuclear myopathy cytoskeleton disease Nemaline myopathy Zaspopathy Channelopathy Myotonia Myotonia congenita Thomsen disease Neuromyotonia / Isaacs syndrome Paramyotonia congenita Periodic paralysis Hypokalemic Thyrotoxic Hyperkalemic Other Central core disease Mitochondrial myopathy MELAS MERRF KSS PEO General Inflammatory myopathy Congenital myopathy v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins
The limitation of motion in axial and proximal joints suggested a rigid spine syndrome. The patients often have hyperextensibility in distal joints, suggesting the Ehlers-Danlos syndrome (see 130000). ... Voermans et al. (2007) reported a patient with Ehlers-Danlos syndrome (EDS; 606408) who had a homozygous mutation in the TNXB gene (600985.0002). ... History Wiedemann (1991) gave an account of Otto Ullrich (1894-1957) that included information on his teacher, von Pfaundler, his career, his personality, and 'his' syndromes. INHERITANCE - Autosomal recessive - Autosomal dominant GROWTH Weight - Low weight due to poor feeding Other - Failure to thrive - Slender build HEAD & NECK Face - Facial weakness, mild - Round face Ears - Prominent ears Mouth - High-arched palate Neck - Torticollis - Neck weakness RESPIRATORY - Respiratory insufficiency due to muscle weakness often requiring ventilatory assistance - Nocturnal hypoventilation - Recurrent chest infections SKELETAL - Proximal joint contractures - Distal joint laxity Spine - Spinal rigidity - Scoliosis - Kyphosis Pelvis - Hip dislocation Limbs - Long, thin limbs - Increased laxity of wrists - Increased laxity of ankles Hands - Increased laxity of fingers Feet - Talipes equinovarus - Calcaneal protrusion SKIN, NAILS, & HAIR Skin - Hyperhidrosis - Follicular hyperkeratosis MUSCLE, SOFT TISSUES - Hypotonia, neonatal - Muscle weakness, proximal greater than distal - Generalized muscle atrophy - Delayed motor milestones - Delayed ambulation - Some patients never achieve ambulation - Muscle biopsy shows increased variation in fiber size - Muscle biopsy shows type 1 fiber predominance - Muscle biopsy shows merosin ( 156225 )-positive muscle fibers - Absence of collagen VI immunostaining - Increased endo- and perimysial connective tissue - Muscle fiber necrosis - Muscle fiber regeneration NEUROLOGIC Central Nervous System - Normal intelligence Peripheral Nervous System - Decreased or absent reflexes due to muscle weakness LABORATORY ABNORMALITIES - Normal to mildly increased serum creatine kinase MISCELLANEOUS - Onset in infancy - Variable severity - Progressive disorder - Bethlem myopathy ( 158810 ) is an allelic disorder with a milder phenotype and autosomal dominant inheritance - A subset of patients have heterozygous mutations consistent with a dominant-negative effect MOLECULAR BASIS - Caused by mutation in the collagen VI, alpha-1 polypeptide gene (COL6A1, 120220.0007 ) - Caused by mutation in the collagen VI, alpha-2 polypeptide gene (COL6A2, 120240.0002 ) - Caused by mutation in the collagen VI, alpha-3 polypeptide gene (COL6A3, 120250.0002 ) ▲ Close
Differential diagnosis In the neonatal period, the differential diagnoses include Bethlem myopathy and other forms of congenitalmuscular dystrophy (CMD) and myopathy, spinal muscular atrophy, forms of Ehlers-Danlos syndrome, and Marfan syndrome (see these terms). Some CMD subtypes such as merosin-deficient congenital muscular dystrophy (MDC1A), Walker-Warburg syndrome, muscle-eye-brain disease, and Fukuyama CMD (see these terms) should also be considered, although in these disorders intellectual deficit is a major symptom.
Ullrich congenital muscular dystrophy is a condition that mainly affects skeletal muscles (the muscles used for movement). Affected individuals show severe muscle weakness soon after birth, develop stiff joints (contractures) in their knees and elbows, and may have an unusual range of movement ( hypermobility ) in their wrists and ankles. This condition is caused by mutations in the COL6A1 , COL6A2 , and COL6A3 genes. Ullrich congenital muscular dystrophy is typically inherited in an autosomal recessive pattern. In rare cases, this condition may be inherited in an autosomal dominant pattern.
A number sign (#) is used with this entry because of evidence that Ullrich congenital muscular dystrophy-2 (UCMD2) is caused by homozygous mutation in the COL12A1 gene (120320) on chromosome 6q. One such family has been reported. For a discussion of genetic heterogeneity of Ullrich congenital muscular dystrophy, see UCMD1 (254090). Clinical Features Zou et al. (2014) reported 2 brothers, born to consanguineous Turkish parents, with features suggesting Ullrich congenital muscular dystrophy. The older brother was profoundly weak at birth, with strikingly hypermobile distal joints, moderate and more proximal joint contractures, and kyphoscoliosis. He also had mild facial weakness, a high-arched palate, and absent deep tendon reflexes.
Ullrich disease Specialty Dermatology Ullrich disease is a genetic extracellular matrix diseases of the skin characterized by puffy skin. [1] See also [ edit ] Ehlers–Danlos syndrome List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
Vasculitis represents a clinically heterogenous group of diseases of multifactorial etiology characterized by inflammation of either large-sized vessels (large-vessel vasculitis, e.g. Giant-cell arteritis and Takayasu arteritis; see these terms), medium-sized vessels (medium-vessel vasculitis e.g. polyarteritis nodosa and Kawasaki disease; see these terms), or small-sized vessels (small-vessel vasculitis, e.g. granulomatosis with polyangiitis, microscopic polyangiitis, immunoglobulin A vasculitis, and cutaneous leukocytoclastic angiitis; see these terms). Vasculitis occurs at any age, may be acute or chronic, and manifests with general symptoms such as fever, weight loss and fatigue, as well as more specific clinical signs depending on the type of vessels and organs affected. The degree of severity is variable, ranging from life or sight threatening disease (e.g. Behçet disease, see this term) to relatively minor skin disease.
Overview Vasculitis involves inflammation of the blood vessels. The inflammation can cause the walls of the blood vessels to thicken, which reduces the width of the passageway through the vessel. If blood flow is restricted, it can result in organ and tissue damage. There are many types of vasculitis, and most of them are rare. Vasculitis might affect just one organ, or several. The condition can be short term or long lasting. Vasculitis can affect anyone, though some types are more common among certain age groups. Depending on the type you have, you may improve without treatment. Most types require medications to control the inflammation and prevent flare-ups.
Summary Clinical characteristics. Myotonia congenita is characterized by muscle stiffness present from childhood; all striated muscle groups including the extrinsic eye muscles, facial muscles, and tongue may be involved. Stiffness is relieved by repeated contractions of the muscle (the "warm-up" phenomenon). Muscles are usually hypertrophic. The autosomal recessive form of myotonia congenita is often associated with more severe symptoms than the autosomal dominant form. Individuals with the autosomal recessive form may have progressive, minor distal weakness and attacks of transient weakness brought on by movement after rest. The age of onset is variable: in autosomal dominant myotonia congenita, onset of symptoms is usually in infancy or early childhood; in the autosomal recessive form, the average age of onset is slightly older.
A rare, genetic, skeletal muscle channelopathy characterized by slow muscle relaxation after contraction (myotonia). Epidemiology Worldwide prevalence is estimated at 1/100,000. Clinical description Onset occurs early in life, usually in the first two decades, with myotonia potentially affecting every muscle after contraction, most frequently lower limb and hand muscles. Myotonia usually improves with exercise (e.g. after warm-up). Myotonia congenita may be inherited as an autosomal dominant (Thomsen disease) or recessive (Becker's disease) inheritance, with a more severe and earlier phenotype in recessively inherited disease. Etiology Disease is caused by loss of function mutations in the gene encoding the chloride channel, CLCN1 (7q34), that plays a role in muscle cell repolarization. Diagnostic methods The clinical diagnosis can easily be confirmed by electromyography (EMG), which reveals myotonic discharges in association with hyperexcitation of the muscle fiber membrane.
Batten (1910) and later Turner (1949) and Turner and Lees (1962) provided 50 years' observations on a family in which 6 sibs presented in infancy the picture of 'amyotonia congenita' and later in life a nonprogressive myopathy. The parents were not related. Muscle - Congenital myopathy Neuro - Amyotonia congenita - Nonprogressive myopathy Inheritance - Autosomal recessive ▲ Close
Diseased mucous membranes can cause constipation and make it hard to eat. Kindler syndrome. This type tends to cause blisters in multiple layers and so can look very different from person to person.
In people with EB, blisters form in response to minor injuries or friction, such as rubbing or scratching.[2310] There are four main types of EB, which are classified based on the depth, or level, of blister formation: Epidermolysis bullosa simplex Dystrophic epidermolysis bullosa Junctional epidermolysis bullosa Kindler Syndrome EB may then be further classified based on severity and specific symptoms, such as distribution (localized or generalized) and whether parts of the body other than the skin are affected.
The spectrum of CDC73 -related disorders includes the following phenotypes: Hyperparathyroidism-jaw tumor (HPT-JT) syndrome. Primary hyperparathyroidism, the main finding of HPT-JT syndrome, occurs in up to 95% of affected individuals; onset is typically in late adolescence or early adulthood. ... Primary hyperparathyroidism occurs in up to 95% of individuals with HPT-JT syndrome; the onset is typically in late adolescence or early adulthood. ... In approximately 10%-15% of individuals with HPT-JT syndrome, primary hyperparathyroidism is caused by parathyroid carcinoma. ... It is not clear whether juvenile fibromas are part of HPT-JT syndrome. Of note, the jaw tumors of HPT-JT syndrome are distinct from the "brown" tumors associated with severe hyperparathyroidism (osteitis fibrosa cystica) and do not resolve following curative parathyroidectomy. ... In 2009 Iacobone et al evaluated 17 individuals with HPT-JT syndrome from three large families; 82% of individuals had single gland disease.
The diagnosis of the 7q11.23 duplication syndrome is established by detection of a recurrent 1.5- to 1.8-Mb heterozygous duplication of the Williams-Beuren syndrome critical region (WBSCR). ... Genetic counseling. The 7q11.23 duplication syndrome is transmitted in an autosomal dominant manner. ... Establishing the Diagnosis The diagnosis of the 7q11.23 duplication syndrome is established by detection of a 1.5- to 1.8-Mb heterozygous duplication of the Williams-Beuren syndrome critical region (WBSCR) (Table 1). ... Differential Diagnosis The 7q11.23 duplication syndrome should be distinguished from other syndromes that include developmental delay, macrocephaly, hypotonia, distinctive craniofacies, and behavior problems. Examples include fragile X syndrome and Sotos syndrome. The 7q11.23 duplication syndrome should be added to the list of syndromes that are associated with aortic dilation: Marfan syndrome, Loeys-Dietz syndrome, Ehlers-Danlos syndromes (see Ehlers-Danlos Syndrome, Classic Type, Ehlers-Danlos Syndrome, Hypermobility Type, Ehlers-Danlos Syndrome, Kyphoscoliotic Form, and EDS vascular type), and familial thoracic aneurysm.
Description The MECP2 gene is mutated in Rett syndrome (RTT; 312750), a severe neurodevelopmental disorder that almost always occurs in females. Although it was first thought that MECP2 mutations causing Rett syndrome were lethal in males, later reports identified a severe neonatal encephalopathy in surviving male sibs of patients with Rett syndrome. ... Zeev et al. (2002) reported an Israeli family in which a girl had classic Rett syndrome and her brother had severe neonatal encephalopathy. ... His sister and aunt, who both carried the mutation, had Rett syndrome. His carrier mother had motor coordination problems, mild learning disability, and skewed X inactivation. Villard et al. (2000) reported a family in which a daughter had classic Rett syndrome and her 2 brothers died in infancy from severe encephalopathy.
Severe neonatal-onset encephalopathy with microcephaly is a rare monogenic disease with epilepsy characterized by neonatal-onset encephalopathy, microcephaly, severe developmental delay or absent development, breathing abnormalities (including central hypoventilation and/or respiratory insufficiency), intractable seizures, abnormal muscle tone and involuntary movements. Early death is usual.
MECP2 -related severe neonatal encephalopathy is the most severe condition in a spectrum of disorders with the same genetic cause. The mildest is PPM-X syndrome, followed by MECP2 duplication syndrome, then Rett syndrome (which exclusively affects females), and finally MECP2 -related severe neonatal encephalopathy. ... Instead, they typically develop Rett syndrome, which has signs and symptoms that include intellectual disability, seizures, and movement problems.