Mena et al. (1991) concluded that the findings, although similar to those in the Fraser syndrome (219000), were sufficiently different to justify recognition as a distinct entity.
Gianotti Crosti syndrome (GCS) is a rare childhood skin condition characterized by a papular rash with blisters on the skin of the legs, buttocks, and arms.
Epidemiology [ edit ] Bogart–Bacall syndrome can develop in individuals at any age. ... Women are more likely than men to develop BBS due to the tendency of lowering their voices in a professional environment. This syndrome is also more prevalent in the 40–50-year-old-age group as their vocal cords thin. ... ISBN 9781597566445 . ^ "RightDiagnosis: Bogart-Bacall syndrome" . Retrieved 2011-09-03 . ^ Koufman, James A.; Blalock, P. ... "Vocal fatigue and dysphonia in the professional voice user: Bogart-bacall syndrome". The Laryngoscope . 98 (5): 493–498. doi : 10.1288/00005537-198805000-00003 . ^ "Natural Health Care - Singers and Musicians" . ... Retrieved 17 December 2020 . ^ "Bogart-Bacall Syndrome" . YouTube . Medical Centric .
PPSH can be difficult to distinguish from the incomplete testicular feminization syndrome (PAIS; 312300), also known as Reifenstein syndrome, especially in the young child. The distinction is obviously important since PPSH is a male-limited autosomal recessive with a recurrence risk of 1 in 8, whereas PAIS is X-linked recessive as is the complete syndrome (AIS; 300068). Wilson et al. (1974) chose to refer to PPSH as type 2 familial incomplete male pseudohermaphroditism, type 1 being Reifenstein syndrome. ... Clinical Management Price et al. (1984) presented evidence that high dose androgen therapy may improve virilization, self-image, and sexual performance in patients with alpha-reductase deficiency who have male-gender behavior and in those patients with Reifenstein syndrome (312100) who have normal amounts of a qualitatively abnormal androgen receptor.
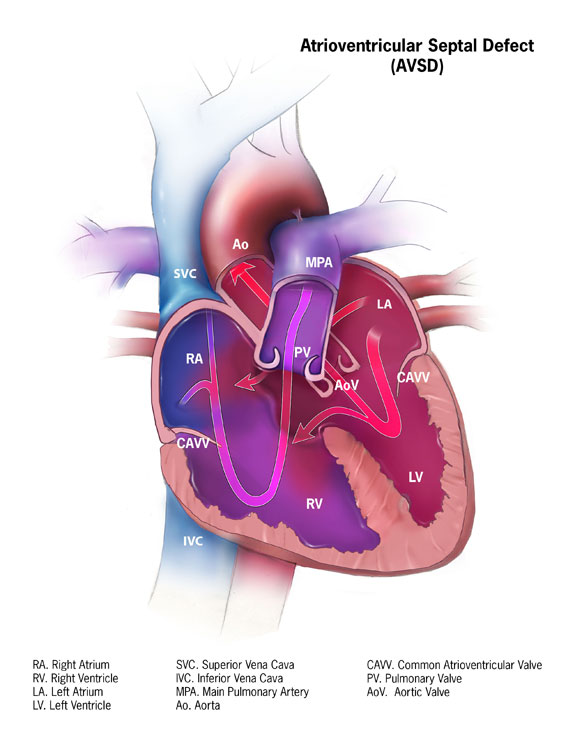
When this happens, the pressure backs up into the pulmonary veins and the lungs. [1] This type of damage is irreversible which is why immediate treatment is recommended after diagnosis. [2] Associated conditions [ edit ] Down syndrome is often associated with AVCD. [3] Other risk factors include: having a parent with a congenital heart defect , alcohol use while pregnant, uncontrolled diabetes treatment during pregnancy and some medications during pregnancy. [1] This type of congenital heart defect is associated with patients with Down syndrome (trisomy 21) or heterotaxy syndromes . [4] 45% of children with Down syndrome have congenital heart disease. Of these, 35–40% have AV septal defects. [5] Similarly, one-third of all children born with AVSDs also have Down syndrome. [6] A study also showed that there is also an increased risk of atrioventricular canal in patients who suffer from Noonan syndrome . The pattern seen in those patients with Noonan syndrome differ from those patients who have Down syndrome in that "partial" AVCD is more prevalent in those who suffer from NS, where as those who suffer from down syndrome show a prevalence of the "complete" form of AVCD. [7] Pathophysiology [ edit ] If there is a defect in the septum, it is possible for blood to travel from the left side of the heart to the right side of the heart, or the other way around. ... "Cardiovascular abnormalities in Down's syndrome: spectrum, management and survival over 22 years" . ... "Complete atrioventricular septal defect, Down syndrome and surgical outcome: Risk factors".
A rare congenital malformation syndrome characterized by the association of facial and skeletal anomalies with severe intellectual deficit and occasional genitourinary anomalies.
Clinical Features Richieri-Costa et al. (1985) described an apparently 'new' autosomal recessive MCA/MR syndrome in a boy and girl, offspring of first-cousin parents. ... Prontera et al. (2011) described 2 Indian sisters with a multiple congenital anomalies/intellectual disability syndrome consistent with AFFND1. At birth, the proband showed bilateral cleft lip/palate, minor facial anomalies, bilateral complete syndactyly of the third and fourth digits, and clubfeet. ... Inheritance The occurrence of this syndrome in sibs and in consanguineous families suggests autosomal recessive inheritance (Guion-Almeida and Richieri-Costa, 2003).
Clinical Features Chudley et al. (1999) reported a family in which 3 males spanning 2 generations had a syndromic form of moderate mental retardation with seizures and dysmorphic facial features. ... Mapping Using microsatellite markers on the X chromosome to evaluate a family with X-linked syndromic mental retardation, Chudley et al. (1999) found linkage to a region on chromosome Xq21.33-q23 between DXS1170 and DXS8067 (maximum 2-point lod score of 2.23 at DXS1120).
Molecular Genetics TGCT Locus and Klinefelter Syndrome Rapley et al. (2000) pointed out that Klinefelter syndrome (47,XXY) is a risk factor for extragonadal germ cell tumors. The relative risk of mediastinal germ cell tumors in Klinefelter syndrome is 67 (Hasle et al., 1995), and 8% of males with mediastinal germ cell tumors have Klinefelter syndrome (Hasle et al., 1992). This raised the possibility that 2 active normal copies of a putative TGCT1 gene may be responsible for the increased risk of germ cell tumors in Klinefelter syndrome.
Genetics [ edit ] This condition may be part of a more complex syndrome or an isolated mutation. Isolated cases are due to mutations in the lipase member H ( LIPH ), lysophosphatidic acid receptor 6 ( LPAR6 ) or keratin 2A ( KRT2 ) genes. ... Differential diagnosis [ edit ] Cardiofaciocutaneous syndrome Naxos disease Palmoplantar keratoderma and cardiomyopathy syndrome [2] Treatment [ edit ] There is no treatment for this condition known at present. Prognosis [ edit ] In isolate cases life expectancy is normal and there are no other related problems. As part of another syndrome this will depend on the other features of the syndrome.
A number sign (#) is used with this entry because of evidence that Seckel syndrome-6 (SCKL6) can be caused by homozygous mutation in the CEP63 gene (614724) on chromosome 3q22. ... For a general phenotypic description and a discussion of genetic heterogeneity of Seckel syndrome, see SCKL1 (210600). Clinical Features Sir et al. (2011) ascertained a consanguineous Pakistani family in which 3 female cousins were born with microcephaly, with head circumferences of -4 SD to -6 SD.
Differential diagnosis Differential diagnoses include neuropathies of the neighboring nerves (ilio-inguinal, genitofemoral, lower cluneal), coccygodynia (given the location of pain projecting into the anus and rectum, aggravated by sitting position) and myofascial syndromes of the deep gluteus muscles (piriformes, obturator internus muscle, levator ani). ... Isolated chronic urethralgia or bladder pain syndrome may be considered when perineal pain varies with urination.
Pudendal neuralgia occurs when the pudendal nerve is injured, irritated, or compressed. Symptoms include burning pain (often unilateral), tingling, or numbness in any of the following areas: buttocks, genitals, or perineum (area between the buttocks and genitals). Symptoms are typically present when a person is sitting but often go away when the person is standing or lying down. The pain tends to increase as the day progresses. Additional symptoms include pain during sex and needing to urinate frequently and/or urgently. Damage to the pudendal nerve can result from surgical procedures, childbirth, trauma, spasms of the pelvic floor muscles, or tumors.
Clinical Features Gokce et al. (2001) reported a 22-year-old female with features of Marfan syndrome (154700) combined with abdominal situs inversus, dextrocardia, and polysplenia. ... Gokce et al. (2001) found no previously reported case of Marfan syndrome combined with complete situs inversus or discrete subaortic stenosis. ... The boy's parents were phenotypically normal; both of the girl's parents were tall and had mitral valve prolapse without other Marfan syndrome stigmata. Both children had dextrocardia and abdominal situs inversus, obstructive sleep apnea, severe scoliosis, and normal chromosome analyses. Physical examinations in both showed signs of Marfan syndrome: pectus carinatum, reduced upper to lower body segment ratio, finger hyperextensibility, and positive wrist sign. ... Kosaki et al. (2004) concluded that these children and the patient reported by Gokce et al. (2001) had a recognizable, distinct syndrome as none fulfilled the Ghent diagnostic criteria for Marfan syndrome and no fibrillin or collagen abnormalities were found.
They concluded that 'with exception of the cases where EA is part of a chromosomal or of a known monogenic or teratogenic syndrome, the recurrence risks fit into a multifactorial scheme.' ... Shaw-Smith (2006) stated that in approximately half of the cases (syndromic esophageal atresia), there are associated anomalies, with cardiac malformations being the most common. ... Molecular Genetics In a review, Shaw-Smith (2006) noted that data from twin and family studies had suggested that genetic factors do not play a major role; there are, however, at least 3 genes that had been identified as playing a role in the etiology of esophageal atresia: NMYC (164840) in Feingold syndrome (164280); SOX2 (184429) in anophthalmia-esophageal-genital syndrome (AEG syndrome, 206900); and CHD7 (608892) in CHARGE syndrome (214800).
Oesophageal atresia (OA) encompasses a group of congenital anomalies with an interruption in the continuity of the oesophagus, with or without persistent communication with the trachea. Epidemiology OA occurs in 1 in 2500 live births. Clinical description Infants with OA are unable to swallow saliva and are noted to have excessive salivation requiring repeated suctioning. Associated anomalies occur in 50% of cases, the majority involving one or more of the VACTERL association anomalies (vertebral, anorectal, cardiac, tracheooesophageal, renal and limb defects). In 86% of cases there is a distal tracheooesophageal fistula, in 7% of cases there is no fistulous connection, while in 4% of cases there is a tracheooesophageal fistula without atresia. The remaining cases are made up of patients with OA with proximal, or both proximal and distal, tracheooesophageal fistula.
Differential diagnosis The differential diagnosis should include other forms of CL (autosomal recessive type 2, autosomal dominant and X-lined CL) and related syndromes (gerodermia osteodysplastica, Cantu syndrome, wrinkly skin syndrome and De Barsy syndrome), together with the Ehlers-Danlos syndromes and Costello syndrome (see these terms).
Cleft velum can disrupt sucking-swallowing in newborns to varying degrees. In the non-syndromic forms, normal breastfeeding is possible. In syndromic forms, there is a risk of food aspiration (isolated or syndromic Pierre Robin sequence). ... Differential diagnosis The presence of associated malformations allows for differentiation between isolated and syndromic forms. Differential diagnoses include syndromic hereditary forms of cleft velum (Pierre-Robin, Stickler, van der Woude and velocardiofacial syndromes;see these terms).
Jenkins and Stady (1980) described a family with simple cleft palate (cleft of the soft palate) in 7 males of 5 sibships in 4 generations. Mouth - Cleft soft palate Inheritance - Autosomal dominant ▲ Close
Two subtypes of IHH have been defined: Kallmann syndrome (CHH with anosmia) mainly associated with abnormal embryonic migration of the Gn-releasing hormone (GnRH)-synthesizing neurons, and normosmic IHH (nIHH), in which HH is the only manifestation, mainly associated with anomalies in the regulation of GnRH signaling and secretion. ... CHH is also a feature of several syndromes including the Prader-Willi, Bardet-Biedl, Laurence-Moon and CHARGE syndromes. ... The following investigations are useful for guiding the molecular diagnosis and for determining the cause of CHH: familial and patient history, tests for anosmia and hearing loss, evaluation for signs associated with dental agenesis and developmental anomalies of the hands and feet, and MRI for identifying olfactory bulb and/or sulcus anomalies and for detecting developmental anomalies of the pituitary or pituitary stalk interruption syndrome. Differential diagnosis Differential diagnoses should include other causes of micropenis and cryptorchidism at birth (syndromic or isolated), transitory HH (associated with constitutional delay of puberty), hypothyroidism, and secondary causes of HH (hypothalamic-pituitary tumors or adenomas, surgical or radiation therapy-induced sequelae, etc.). Genetic counseling Genetic counseling may be proposed depending on the underlying disorder (syndromic or isolated) and mode of transmission (X-linked, or autosomal recessive or dominant).