When it is, it's usually associated with genetic disorders, such as Down syndrome. Risk factors Factors that increase your risk of developing ectropion include: Age.
The rare polydactylies were frequently syndromal: one-third of them (77/236) were found in association with other congenital anomalies, 11% (26/236) in multiple congenital anomaly (MCA) cases, and 21.6% (51/236) in recognized syndromes. ... Associated cases were further subdivided into (1) combined, if the other defect was a limb defect; (2) syndromic, if the non-limb defect constituted a recognized causal or pathogenetic entity; and (3) MCA (multiple congenital anomalies), if a non-limb defect did not constitute a recognized entity. ... Polydactylies were rarely associated with other congenital anomalies except in recognizable syndromes; when syndromes were excluded, most of the significant positive associations disappeared. Trisomy 13, Meckel syndrome (249000), and Down syndrome (190685) explained 255 of the 338 syndromic polydactyly cases. Down syndrome was strongly associated with first-digit duplication, and negatively associated with postaxial polydactyly.
However, it can occur in association with other birth defects and cognitive abnormalities as part of a genetic syndrome. In some cases, the extra digits may be well-formed and functional.
You can help Wikipedia by expanding it . v t e v t e Symptoms and signs relating to the nervous system Neurological examination · Cranial nerve examination Central nervous system Head Battle's sign Kernig's sign Macewen's sign Myerson's sign Stroop test Hirano body Other increased intracranial pressure Cushing's triad Lhermitte's sign Charcot's neurologic triad Peripheral nervous system Reflexes Combination Jendrassik maneuver Legs Plantar reflex Chaddock reflex Oppenheim's sign Westphal's sign Arms Hoffmann's sign Other Arms Froment's sign carpal tunnel syndrome Tinel sign Phalen maneuver Legs Gowers' sign Hoover's sign Lasègue's sign Trendelenburg's sign Torso Beevor's sign General Pain stimulus v t e Diseases relating to the peripheral nervous system Mononeuropathy Arm median nerve Carpal tunnel syndrome Ape hand deformity ulnar nerve Ulnar nerve entrapment Froment's sign Ulnar tunnel syndrome Ulnar claw radial nerve Radial neuropathy Wrist drop Cheiralgia paresthetica long thoracic nerve Winged scapula Backpack palsy Leg lateral cutaneous nerve of thigh Meralgia paraesthetica tibial nerve Tarsal tunnel syndrome plantar nerve Morton's neuroma superior gluteal nerve Trendelenburg's sign sciatic nerve Piriformis syndrome Cranial nerves See Template:Cranial nerve disease Polyneuropathy and Polyradiculoneuropathy HMSN Charcot–Marie–Tooth disease Dejerine–Sottas disease Refsum's disease Hereditary spastic paraplegia Hereditary neuropathy with liability to pressure palsy Familial amyloid neuropathy Autoimmune and demyelinating disease Guillain–Barré syndrome Chronic inflammatory demyelinating polyneuropathy Radiculopathy and plexopathy Brachial plexus injury Thoracic outlet syndrome Phantom limb Other Alcoholic polyneuropathy Other General Complex regional pain syndrome Mononeuritis multiplex Peripheral neuropathy Neuralgia Nerve compression syndrome v t e Musculoskeletal examination Leg Hip examination Galeazzi test Allis test Barlow maneuver Ober's test Ortolani test Patrick's test Thomas test Trendelenburg's sign Knee examination Ballottement Clarke's test Drawer test Lachman test Patellar tap Pivot-shift test Valgus stress test meniscus Apley grind test McMurray test ligament and meniscus Unhappy triad Foot and ankle Hubscher's maneuver Mulder's sign Simmonds' test Thompson test Ankle Simmonds' test General Straight leg raise Lasègue's sign Gait abnormality Trendelenburg gait Unequal leg length Arm Shoulder examination Apprehension test Jobe's test Neer impingement sign Yergason's test rotator cuff Hawkins–Kennedy test Watson's test Elbow examination Cozen's test Elbow extension test Hand and wrist Durkan's test Finkelstein's test Froment's sign Lunotriquetral shear test Phalen maneuver Tinel sign Watson's test Spine Gaenslen's test Low back pain Waddell's signs Lower back flexibility Schober's test sacroiliitis Larrey's sign Other Range of motion Palpation Codman triangle
In addition, Biofeedback and physical therapy are used to strengthen the muscles. [3] See also [ edit ] Friedrich Trendelenburg Gait abnormality Gluteal gait Snapping hip syndrome Trendelenburg position Trendelenburg's sign References [ edit ] ^ Viraj N. ... Wheeless' textbook of orthopaedics [1] Ropper and Brown, Adams and Victor's Principles of Neurology , 8th edition (2005), p. 105 External links [ edit ] Classification D DiseasesDB : 29422 hip/hip%20clin%20correl/corr6 at the Dartmouth Medical School 's Department of Anatomy v t e Symptoms and signs relating to movement and gait Gait Gait abnormality CNS Scissor gait Cerebellar ataxia Festinating gait Marche à petit pas Propulsive gait Stomping gait Spastic gait Magnetic gait Truncal ataxia Muscular Myopathic gait Trendelenburg gait Pigeon gait Steppage gait Antalgic gait Coordination Ataxia Cerebellar ataxia Dysmetria Dysdiadochokinesia Pronator drift Dyssynergia Sensory ataxia Asterixis Abnormal movement Athetosis Tremor Fasciculation Fibrillation Posturing Abnormal posturing Opisthotonus Spasm Trismus Cramp Tetany Myokymia Joint locking Paralysis Flaccid paralysis Spastic paraplegia Spastic diplegia Spastic paraplegia Syndromes Monoplegia Diplegia / Paraplegia Hemiplegia Triplegia Tetraplegia / Quadruplegia General causes Upper motor neuron lesion Lower motor neuron lesion Weakness Hemiparesis Other Rachitic rosary Hyperreflexia Clasp-knife response v t e Musculoskeletal examination Leg Hip examination Galeazzi test Allis test Barlow maneuver Ober's test Ortolani test Patrick's test Thomas test Trendelenburg's sign Knee examination Ballottement Clarke's test Drawer test Lachman test Patellar tap Pivot-shift test Valgus stress test meniscus Apley grind test McMurray test ligament and meniscus Unhappy triad Foot and ankle Hubscher's maneuver Mulder's sign Simmonds' test Thompson test Ankle Simmonds' test General Straight leg raise Lasègue's sign Gait abnormality Trendelenburg gait Unequal leg length Arm Shoulder examination Apprehension test Jobe's test Neer impingement sign Yergason's test rotator cuff Hawkins–Kennedy test Watson's test Elbow examination Cozen's test Elbow extension test Hand and wrist Durkan's test Finkelstein's test Froment's sign Lunotriquetral shear test Phalen maneuver Tinel sign Watson's test Spine Gaenslen's test Low back pain Waddell's signs Lower back flexibility Schober's test sacroiliitis Larrey's sign Other Range of motion Palpation Codman triangle
Gluteal gait is an abnormal gait caused by neurological problems. If the superior gluteal nerve or obturator nerves are injured, they fail to control the gluteus minimus and medius muscles properly, thus producing an inability to tilt the pelvis upward while swinging the leg forward to walk. To compensate for this loss, the leg swings out laterally so that the foot can move forward, producing a shuffling or waddling gait. Injury to the superior gluteal nerve results in a characteristic motor loss, resulting in a disabling gluteus medius limp, to compensate for weakened abduction of the thigh by the gluteus medius and minimus, and/or a gluteal gait, a compensatory list of the body to the weakened gluteal side. As a result of this compensation, the center of gravity is placed over the supporting lower limb. Medial rotation of the thigh is also severely impaired. When a person is asked to stand on one leg, the gluteus medius and minimus normally contract as soon as the contralateral foot leaves the floor, preventing tipping of the pelvis to the unsupported side.
Rare disorder linked to overgrowth and is characterized by dysmorphic facial features Cohen-Gibson syndrome Autosomal dominant is the manner in which this condition is inherited Cohen-Gibson Syndrome is a disorder linked to overgrowth and is characterized by dysmorphic facial features and variable intellectual disability. ... It has been reported at least four times in different racial demographics, once in a Turkish, Hispanic, Japanese and Caucasian patient. [1] References [ edit ] ^ a b c "COHEN-GIBSON SYNDROME; COGIS" . Retrieved 21 October 2019 . ^ Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, et al. ... "Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome" . Human Mutation . 38 (6): 637–648. doi : 10.1002/humu.23200 . PMID 28229514 . v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis
A number sign (#) is used with this entry because of evidence that Cohen-Gibson syndrome (COGIS) is caused by heterozygous mutation in the EED gene (605984) on chromosome 11q14. Description Cohen-Gibson syndrome is an overgrowth disorder characterized by increased somatic parameters apparent from birth and associated with variable intellectual disability. ... Clinical Features Cohen et al. (2015) reported a 27-year-old man, born of unrelated Turkish parents, with an overgrowth syndrome associated with intellectual disability.
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome. A form of holoprosencephaly (HPE10) has been mapped within the deleted region of chromosome 1q41-q42. ... See also congenital diaphragmatic hernia (DIH; 142340), which has been associated with deletion of chromosome 1q41-q42. See also Skraban-Deardorff syndrome (SKDEAS; 617616), caused by mutation in the WDR26 gene (617424) on chromosome 1q42, which shows overlapping features with chromosome 1q41-q42 deletion syndrome. ... Two individuals had diaphragmatic hernia and lung hypoplasia with a clinical diagnosis of Fryns syndrome (229850; see, e.g., Van Hove et al., 1995). Shaffer et al. (2007) proposed that the 1q41-q42 deletion syndrome has a variable presentation, with the extreme end of the spectrum demonstrating a Fryns syndrome phenotype. ... Slavotinek et al. (2006) identified a de novo interstitial deletion of 1q32.3-q42.2 in a male with congenital diaphragmatic hernia and pulmonary hypoplasia with multiple other congenital anomalies suggestive of Fryns syndrome. Using array-based comparative genomic hybridization, Shaffer et al. (2007) identified 7 patients with de novo heterozygous microdeletions of chromosome 1q41-q42 that included the DISP1 gene (607502).
A number sign (#) is used with this entry because of evidence that holoprosencephaly-4 (HPE4) is caused by heterozygous mutation in the TGIF gene (602630) on chromosome 18p11. For phenotypic information and a general discussion of genetic heterogeneity in holoprosencephaly, see HPE1 (236100). Cytogenetics Johnson and Bachman (1976) described a normal female who appeared to have a nonreciprocal translocation from the short arm of one chromosome 18 to the long arm of a chromosome 12. She gave birth to a cebocephalic child whose karyotype included an 18p- chromosome. The association of loss of 18p with holoprosencephaly was suggested by the patient reported by Munke et al. (1988); cytogenetic and molecular studies indicated a Y/18 translocation with loss of 18p and distal Yq material in a holoprosencephalic fetus.
A number sign (#) is used with this entry because holoprosencephaly-11 (HPE11) is caused by heterozygous mutation in the CDON gene (608707) on chromosome 11q24. For a general phenotypic description and a discussion of genetic heterogeneity of holoprosencephaly, see HPE1 (236100). Clinical Features Bae et al. (2011) reported 4 unrelated patients with HPE11. One patient had agenesis of the corpus callosum, hypotelorism, growth hormone deficiency, global developmental delay, and thick eyebrows with synophrys. Another had agenesis of the corpus callosum, alobar HPE, hypotelorism, cleft lip/palate, and absent columella; absent pituitary and polysplenia were noted in this patient at autopsy.
HPE3), TGIF , ZIC2 , SIX3 [8] and BOC genes. [9] Although many children with holoprosencephaly have normal chromosomes , specific chromosomal abnormalities have been identified in some patients ( trisomy of chromosome 13 , also known as Patau syndrome ). There is evidence that in some families, HPE is inherited ( autosomal dominant as well as autosomal or X-linked recessive inheritance ). [10] [11] [12] Features consistent with familial transmission of the disease (e.g., a single central maxillary incisor ) should be carefully assessed in parents and family members. [13] Non-genetic factors [ edit ] Numerous possible risk factors have been identified, including gestational diabetes , transplacental infections (the " TORCH complex "), first trimester bleeding , and a history of miscarriage . [6] [14] As well, the disorder is found twice as often in female babies. [14] However, there appears to be no correlation between HPE and maternal age . [14] There is evidence of a correlation between HPE and the use of various drugs classified as being potentially unsafe for pregnant and lactating mothers. ... External links [ edit ] Classification D ICD - 10 : Q04.2 ICD - 9-CM : 742.2 OMIM : 236100 MeSH : D016142 DiseasesDB : 29610 External resources eMedicine : radio/347 GeneReviews : Holoprosencephaly Overview Orphanet : 2162 GeneReview/NIH/UW entry on Holoprosencephaly Overview holoprosencephaly at NINDS What do we know about holoprosencephaly - Genome.gov v t e Congenital malformations and deformations of nervous system Brain Neural tube defect Anencephaly Acephaly Acrania Acalvaria Iniencephaly Encephalocele Chiari malformation Other Microcephaly Congenital hydrocephalus Dandy–Walker syndrome other reduction deformities Holoprosencephaly Lissencephaly Microlissencephaly Pachygyria Hydranencephaly Septo-optic dysplasia Megalencephaly Hemimegalencephaly CNS cyst Porencephaly Schizencephaly Polymicrogyria Bilateral frontoparietal polymicrogyria Spinal cord Neural tube defect Spina bifida Rachischisis Other Currarino syndrome Diastomatomyelia Syringomyelia
For a phenotypic description and a discussion of genetic heterogeneity of holoprosencephaly, see HPE1 (236100). Clinical Features Levin and Surana (1991) described holoprosencephaly in association with an interstitial deletion of chromosome 14q11.1-q13. Parental karyotypes were normal. The white female, born to nonconsanguineous young parents after an uncomplicated pregnancy, showed hypotelorism, lack of nasal bridge, flattened nasal tip with no visible septum, wide midline cleft of lip and hard palate, and ptosis of the left upper eyelid. Axial CT scan of the head was interpreted as showing semilobar holoprosencephaly. The infant died at 8 days of age. Kamnasaran et al. (2005) reported 6 patients with HPE and interstitial deletions on proximal chromosome 14q: 1 had alobar HPE and 5 had lobar HPE.
For phenotypic information and a general discussion of genetic heterogeneity in holoprosencephaly (HPE), see HPE1 (236100). Clinical Features Lehman et al. (2001) described a female infant who survived for 5.5 hours after delivery at 33 weeks' gestation. Autopsy showed a lobar variant of holoprosencephaly. Cytogenetics By cytogenetic analysis in an infant with a lobar variant of holoprosencephaly, Lehman et al. (2001) identified a 2q37.1-q37.3 deletion. This case represented the fourth reported case of HPE associated with partial monosomy 2q37 and the first with an apparently isolated 2q37 deletion. Lehman et al. (2001) suggested that the deleted segment may contain yet another locus, here designated HPE6, which, when disrupted, can lead to brain malformations within the HPE spectrum.
This condition is called nonsyndromic to distinguish it from other types of holoprosencephaly caused by genetic syndromes, chromosome abnormalities, or substances that cause birth defects (teratogens).
A number sign (#) is used with this entry because of evidence that holoprosencephaly-3 (HPE3) is caused by heterozygous mutation in the SHH gene (600725), which encodes the human Sonic hedgehog homolog, on chromosome 7q36. For a phenotypic description and a discussion of genetic heterogeneity of holoprosencephaly, see HPE1 (236100). Clinical Features Berry et al. (1984) and Johnson (1989) provided information on a family (family 2 in Johnson, 1989) in which holoprosencephaly occurred in 2 sibs and their first cousin, who were offspring of parents with a single central maxillary incisor. Johnson (1989) reported a second patient (family 1) with full-blown holoprosencephaly whose mother and sister had only a single central maxillary incisor. Johnson (1989) suggested that holoprosencephaly is a developmental field defect of which the mild forms can be single median incisor, hypotelorism, bifid uvula, or pituitary deficiency.
One of the patients with holoprosencephaly described by Ribeiro et al. (2006) was considered by Guion-Almeida et al. (2007) to have cerebrooculonasal syndrome (CONS; 605627); see 601309.0015.
A number sign (#) is used with this entry because of evidence that solitary median maxillary central incisor (SMMCI) and SMMCI syndrome are caused by heterozygous mutation in the Sonic hedgehog gene (SHH; 600725) on chromosome 7q36. ... Hall et al. (1997) described a series of 21 consecutive cases of solitary median maxillary central incisor syndrome seen in the Royal Children's Hospital in Melbourne, Australia, from 1966 to 1997. The spectrum of anomalies and associated features present in these cases, including short stature, choanal atresia, midnasal stenosis, and holoprosencephaly, was described. They pointed out that SMMCI syndrome, previously considered a simple midline defect of the dental lamina, is a possible predictor of holoprosencephalies of varying degree in the proband, members of the proband's family, and in the family's descendants. ... Dolan et al. (1981) and Aughton et al. (1991) reported cases of del(18p) syndrome and single maxillary central incisor. ... INHERITANCE - Autosomal dominant GROWTH Height - Short stature HEAD & NECK Head - Microcephaly Eyes - Hypotelorism Mouth - Prominent midpalatal ridge (torus palatinus) Teeth - Single median maxillary central incisor (SMMCI) RESPIRATORY Nasopharynx - Midnasal stenosis - Choanal atresia - Congenital nasal pyriform aperture stenosis ENDOCRINE FEATURES - Isolated growth hormone deficiency - Hypopituitarism MISCELLANEOUS - SMCCI can be an isolated anomaly, part of a syndrome or association as in VACTERL ( 192350 ) and CHARGE ( 214800 ), part of autosomal dominant holoprosencephaly spectrum such as in HPE3 ( 142945 ), HPE2 ( 157170 ), and HPE4 ( 142946 ), or a feature in chromosomal abnormalities such as del(18p) and del(7)(q36->qter) MOLECULAR BASIS - Caused by mutation in the sonic hedgehog gene (SHH, 600725.0014 ) ▲ Close
When holoprosencephaly is combined with severe facial anomalies and postaxial polydactyly, the pseudotrisomy 13 syndrome (264480) should be considered. ... Eleven (37%) of the cases had detectable chromosomal aberrations, and 7 (23%) were suspected to be of nonchromosomal syndromal origin. Chan et al. (2009) described the occurrence of semilobar holoprosencephaly in the child of a mother with mesiodens and suggested that the supernumerary maxillary tooth (see 187100) may be a microform of HPE. ... However, SIM2 did map within the Down syndrome critical region and thus was a candidate gene for contributing to the Down syndrome phenotype. ... Genetic factors are indicated by familial occurrence, the occurrence of holoprosencephaly in some mendelian genetic syndromes, and the association with nonrandom chromosomal aberrations. ... Another 31% of the cases of cyclopias were associated with defects not typically related to HPE, with more hydrocephalus, heterotaxia defects, neural tube defects, and preaxial reduction defects than the chromosomal group, suggesting the presence of ciliopathies or other unrecognized syndromes. Cyclopia is a very rare defect without much variability in prevalence by geographic location.
HPE may also be associated with certain syndromes or chromosomal abnormalities (such as Smith-Lemli-Opitz syndrome, Hartsfield Syndrome and trisomy 13). ... Differential diagnosis Differential diagnosis includes anencephaly, severe congenital hydrocephalus, Walker-Warburg syndrome, large interhemispheric cyst, otocephaly and other midline defects, as well as syndromic and chromosomal-related forms. ... If a genetic etiology is established in a proband, specific counseling for recurrence risk is indicated. In non-syndromic HPE, all modes of inheritance have been described; however, most cases are complex with digenic or oligogenic inheritance.
Hefner used the terms "short thumb" and "brachymegalodactylism" in 1924, [3] and "short thumb" has continued to be used in a few other studies since then, including the study that defined Rubinstein–Taybi syndrome in 1963. [1] "Stub thumb" is the common term preferred by the online database Online Mendelian Inheritance in Man [6] and was first used in a 1965 study. [7] Stub thumbs have also been called murderer's thumb (allegedly among fortune tellers ), [7] bohemian thumb, Tory's thumb, and potter's thumb. [6] The term "clubbed thumb" should not be confused with nail clubbing , which is a clinical sign associated with a number of diseases. ... "Broad Thumbs and Toes and Facial Abnormalities: A Possible Mental Retardation Syndrome". American Journal of Diseases of Children . 105 (6): 588–608. doi : 10.1001/archpedi.1963.02080040590010 . ... Retrieved 2019-08-17 . ^ Williams, Kimberly D. (2013-09-10). "Non-Syndromic Brachydactyly Type D and Type E Mapped to 7p15 in Healthy Children and Adults from the Jirel Ethnic Group in Eastern Nepal" . ... PMID 24022874 . v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum
Sudden acquired retinal degeneration syndrome ( SARDS ) is a disease in dogs causing sudden blindness. ... "Evaluation of a comparative pathogenesis between cancer-associated retinopathy in humans and sudden acquired retinal degeneration syndrome in dogs via diagnostic imaging and western blot analysis". ... "Diagnostic utility of clinical and laboratory test parameters for differentiating between sudden acquired retinal degeneration syndrome and pituitary-dependent hyperadrenocorticism in dogs". ... "Photoreceptor cell death by apoptosis in dogs with sudden acquired retinal degeneration syndrome". Am J Vet Res . 59 (2): 149–52. ... "Sudden acquired retinal degeneration syndrome (SARDS) - a review and proposed strategies toward a better understanding of pathogenesis, early diagnosis, and therapy" (PDF) .
Jumping Frenchmen syndrome shares some symptoms with other startle disorders. ... Also, instances of many being shy may imply that the "jumper" was positively reinforced by the sudden attention as the entertainment for a group. [4] In 1885, Georges Gilles de la Tourette included Jumping Frenchmen syndrome in the typology of "convulsive tic illness"; [7] studies of the condition in the 1980s cast doubt on whether the phenomenon was in fact a physical condition similar to Tourette syndrome . ... Tourette syndrome is characterized by multiple physical (motor) tics and at least one vocal (phonic) tic. There are many overlaps when compared clinically, but the abnormal "jumping" response is always provoked, unlike the involuntary tics in Tourette syndrome. [9] Similar conditions [ edit ] Latah is a disorder found in southeast Asia in which one's startle response is similar to a state of trance with repetitive speech or movements. ... A cursing brain?: The histories of Tourette syndrome . Harvard University Press, 2000.
History The reports by Beard (1878, 1880, 1886) stimulated Georges Gilles de la Tourette to study patients making peculiar sounds and movements and led to description of the disorder his mentor Charcot referred to as Gilles de la Tourette syndrome (137580). Dr. Victor McKusick grew up in the Moosehead Lake region of Maine.
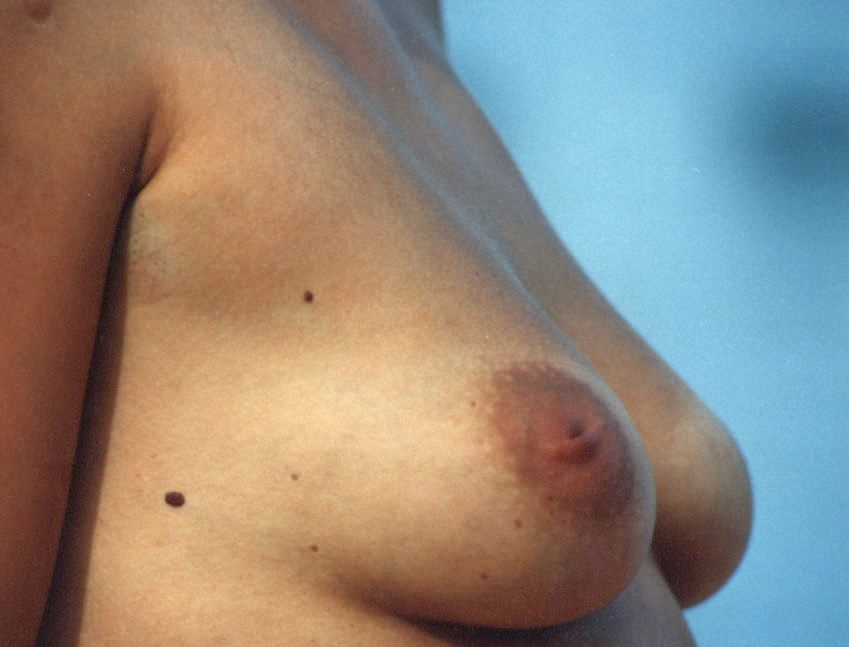
Duct ectasia syndrome in the classical meaning is associated with additional histological changes. [ citation needed ] Contents 1 Symptoms 2 Causes 3 Pathogenesis 4 Diagnosis 5 Duct Ectasia Syndrome 6 Prognosis 7 Terminology 8 References 9 External links Symptoms [ edit ] Signs of duct ectasia can include nipple retraction, inversion, pain, [4] and classic green-brown discharge. ... Smokers seem more often affected by duct ectasia syndrome although the reported results are not entirely consistent. ... Both duct widening and duct ectasia syndrome are frequently bilateral, hence systemic causes are likely involved. ... In plasma cell rich lesions diagnosed on core biopsies, steroid-responsive IgG4-related mastitis can be identified by IgG/IgG4 immunostaining. [6] Duct Ectasia Syndrome [ edit ] The term duct ectasia syndrome has been used to describe symptoms of nonpuerperal mastitis, possibly associated with nipple inversion and nipple discharge. ... Duct ectasia syndrome has been associated with histopathological findings that are distinct from a simple duct widening.
Overview Mammary duct ectasia (ek-TAY-zhuh) occurs when one or more milk ducts beneath your nipple widens. The duct walls may thicken, and the duct may fill with fluid. The milk duct may become blocked or clogged with a thick, sticky substance. The condition often causes no symptoms, but some women may have nipple discharge, breast tenderness or inflammation of the clogged duct (periductal mastitis). Mammary duct ectasia most often occurs in women during perimenopause — around age 45 to 55 years — but it can happen after menopause, too. The condition often improves without treatment. If symptoms persist, you may need antibiotics or possibly surgery to remove the affected milk duct.
This article may be confusing or unclear to readers . Please help us clarify the article . There might be a discussion about this on the talk page . ( November 2020 ) ( Learn how and when to remove this template message ) Hook nail is a bowing of the nail bed due to a lack of support from the short bony phalanx (fingertip). [1] : 660 See also [ edit ] Half and half nails List of cutaneous conditions References [ edit ] ^ Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine . (6th ed.). McGraw-Hill. ISBN 0-07-138076-0 . This condition of the skin appendages article is a stub . You can help Wikipedia by expanding it . v t e
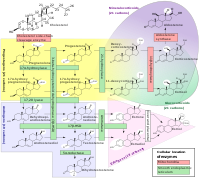
Congenital adrenal hyperplasia (CAH) due to 11-beta-hydroxylase deficiency is one of a group of disorders (collectively called congenital adrenal hyperplasia) that affect the adrenal glands. The adrenal glands are located on top of the kidneys and produce a variety of hormones that regulate many essential functions in the body. In people with CAH due to 11-beta-hydroxylase deficiency, the adrenal glands produce excess androgens, which are male sex hormones. There are two types of CAH due to 11-beta-hydroxylase deficiency, the classic form and the non-classic form. The classic form is the more severe of the two types. Females with the classic form of CAH due to 11-beta-hydroxylase deficiency have external genitalia that do not look clearly male or female (atypical genitalia).
A rare form of congenital adrenal hyperplasia (CAH) characterized by glucocorticoid deficiency, hyperandrogenism, hypertension and virilization in females. Epidemiology It accounts for approximately 5-8% of CAH cases and has an annual incidence of 1/100,000-200,000 live births. Clinical description If the disorder is not recognized during the neonatal period, both girls and boys undergo rapid postnatal growth with accelerated growth velocity and accelerated skeletal maturation (leading to short stature in adulthood) and sexual precocity. Severe virilization is seen in the external genitalia of girls while boys appear normal. Precocious pseudopuberty and hypertension are seen in both sexes. There is also a life-long risk for an adrenal crisis.
A number sign (#) is used with this entry because congenital adrenal hyperplasia (CAH) due to 11-beta-hydroxylase deficiency is caused by homozygous or compound heterozygous mutation in the CYP11B1 gene (610613) on chromosome 8q24. Description Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency is an autosomal recessive disorder of corticosteroid biosynthesis resulting in androgen excess, virilization, and hypertension. The defect causes decreased synthesis of cortisol and corticosterone in the zona fasciculata of the adrenal gland, resulting in accumulation of the precursors 11-deoxycortisol and 11-deoxycorticosterone; the latter is a potent salt-retaining mineralocorticoid that leads to arterial hypertension (White et al., 1991). CAH due to 11-beta-hydroxylase deficiency accounts for approximately 5 to 8% of all CAH cases; approximately 90% of cases are caused by 21-hydroxylase deficiency (201910) (White et al., 1991). Clinical Features The nature of the defect in congenital adrenal hyperplasia associated with hypertension was first demonstrated by Eberlein and Bongiovanni (1956) on the basis of the accumulated steroids.
Congenital adrenal hyperplasia (CAH) due to 11-beta-hydroxylase deficiency is one of a group of disorders (collectively called congenital adrenal hyperplasia ) that affect the adrenal glands . In this condition, the adrenal glands produce excess androgens (male sex hormones). This condition is caused by mutations in the CYP11B1 gene and is inherited in an autosomal recessive pattern. There are two types, the classic form and the non-classic form. Females with the classic form have ambiguous external genitalia with normal internal reproductive organs. Males and females with the classic form have early development of their secondary sexual characteristics ( precocious puberty ).