Bodily reaction Cytokine release syndrome Other names Infusion-related reaction (IRR), infusion reaction, [1] cytokine storm [2] Specialty Immunology Cytokine release syndrome ( CRS ) is a form of systemic inflammatory response syndrome (SIRS) that can be triggered by a variety of factors such as infections and certain drugs. [3] It refers to cytokine storm syndromes (CSS) [4] and occurs when large numbers of white blood cells are activated and release inflammatory cytokines , which in turn activate yet more white blood cells. ... "In vitro cytokine release assays for predicting cytokine release syndrome: the current state-of-the-science. ... PMID 29907163 . ^ Behrens EM, Koretzky GA (June 2017). "Review: Cytokine Storm Syndrome: Looking Toward the Precision Medicine Era". ... "Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy" . ... "GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts" .
Patients who are suffering from Stein-Leventhal syndrome (also referred to as polycystic ovary syndrome, or PCOS) can also suffer from anovulation. [3] Up to 90% of cases of anovulation are caused by PCOS ; this syndrome is usually hereditary . [4] [5] Weight loss or anorexia can also cause hormonal imbalance, leading to irregular ovulation (dysovulation). ... "A 27-year-old woman with a diagnosis of polycystic ovary syndrome". JAMA . 297 (5): 509–519. doi : 10.1001/jama.297.5.509 . ... "The prevalence and features of the polycystic ovary syndrome in an unselected population" . ... Diagnosis and Management of Polycystic Ovary Syndrome . Springer. p. 243. ISBN 978-0-387-09717-6 . ... "PCOSMIC: a multi-centre randomized trial in women with PolyCystic Ovary Syndrome evaluating Metformin for Infertility with Clomiphene" .
Fibrous Dysplasia/McCune-Albright Syndrome . Seattle (WA): University of Washington, Seattle. ... "Polyostotic fibrous dysplasia associated with intramuscular myxomas: Mazabraud's syndrome" . Skeletal Radiology . 27 (5): 278–282. doi : 10.1007/s002560050381 . ... "Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome" . Journal of Bone and Mineral Research . 19 (4): 571–577. doi : 10.1359/JBMR.0301262 . ... "Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation" . ... "Optic neuropathy in McCune-Albright syndrome: effects of early diagnosis and treatment of growth hormone excess" .
This condition can occur alone or as part of a genetic disorder , such as McCune-Albright syndrome . While there is no cure for fibrous dysplasia, the symptoms can be treated.
Hyperthyroidism and growth hormone hypersecretion are also quite frequently observed, while Cushing syndrome is exceptional. These patients also have café-au-lait cutaneous spots. These endocrine or cutaneous features represent the McCune-Albright syndrome. Pancreatic intraductal papillary mucinous neoplasms have been described in patients with extended forms.
Description Restless legs syndrome (RLS) is a neurologic sleep/wake disorder characterized by uncomfortable and unpleasant sensations in the legs that appear at rest, usually at night, inducing an irresistible desire to move the legs. ... For additional information and a discussion of genetic heterogeneity of restless legs syndrome, see RLS1 (102300). Mapping Pichler et al. (2006) reported a genomewide linkage analysis of patients with RLS collected from 1 small village that had conserved a high degree of isolation in the Western Alps of South Tyrol (Italy).
Anonychia congenita can also be part of syndromes that affect multiple parts of the body, including Coffin-Siris syndrome and nail-patella syndrome. When anonychia congenita is part of a syndrome, it is caused by mutations in the gene associated with that syndrome.
A new leukoencephalopathy, the CACH syndrome (Childhood Ataxia with Central nervous system Hypomyelination) or VWM (Vanishing White Matter) was identified on clinical and MRI criteria. Classically, this disease is characterized by (1) an onset between 2 and 5 years of age, with a cerebello-spastic syndrome exacerbated by episodes of fever or head trauma leading to death after 5 to 10 years of disease evolution, (2) a diffuse involvement of the white matter on cerebral MRI with a CSF-like signal intensity (cavitation), (3) a recessive autosomal mode of inheritance, (4) neuropathologic findings consistent with a cavitating orthochromatic leukodystrophy with increased number of oligodendrocytes with sometimes ``foamy'' aspect.
While the juvenile and adult forms are often associated with primary or secondary ovarian failure – a syndrome referred to as "ovarioleukodystrophy" [Schiffmann et al 1997, Fogli et al 2003], ovarian failure may occur in any of the forms regardless of age of onset; it has been found at autopsy in infantile and childhood cases [van der Knaap et al 2003].
Leukoencephalopathy with vanishing white matter is a progressive disorder that mainly affects the central nervous system (CNS). This disorder causes deterioration of white matter , which consists of nerve fibers covered by myelin (the substance that protects the nerves). Most affected people begin to have signs and symptoms during childhood, but symptoms may first become apparent anywhere from before birth to adulthood. Symptoms may include difficulty coordinating movements (ataxia); muscle stiffness (spasticity); and optic atrophy . Symptoms may worsen rapidly with episodes of fever, after head trauma, or with other stresses on the body.
Only 1 patient presented with rapid cognitive decline, including a frontal lobe syndrome. The age at menarche was normal in the 5 patients with secondary amenorrhea. ... Tedeschi et al. (1995) concluded that this syndrome is secondary to a metabolic defect causing hypomyelination, axonal degeneration, and, in the most compromised cases, accumulation of lactate.
Leukoencephalopathy with vanishing white matter is a progressive disorder that mainly affects the brain and spinal cord (central nervous system). This disorder causes deterioration of the central nervous system's white matter, which consists of nerve fibers covered by myelin. Myelin is the fatty substance that insulates and protects nerves. In most cases, people with leukoencephalopathy with vanishing white matter show no signs or symptoms of the disorder at birth. Affected children may have slightly delayed development of motor skills such as crawling or walking. During early childhood, most affected individuals begin to develop motor symptoms, including abnormal muscle stiffness (spasticity) and difficulty with coordinating movements (ataxia).
A number sign (#) is used with this entry because of evidence that Simpson-Golabi-Behmel syndrome type 2 (SGBS2) is caused by mutation in the OFD1 gene (300170) on chromosome Xp22. One such family has been reported. See also orofaciodigital syndrome type I (OFD1; 311200), an allelic disorder with a different phenotype.
PMID 18611176 . ^ a b Pong, Anthony Ham (June 2000). "Oral Allergy Syndrome". Allergy/Asthma Information Association (AAIA) Newsletter . ^ "Oral Allergy Syndrome" . oralallergy.net . ... (April 28, 2007). "Oral Allergy Syndrome" . About.com . Retrieved 2008-01-25 . ^ "OAS Food Allergens" . ... Retrieved 2010-01-26 . ^ "Oral Allergy Syndrome" . Asthma and Allergy Foundation of America . ... -M.; Van Ree, R.; Ceuppens, J.L. (2003). "Oral Allergy Syndrome to Chicory Associated with Birch Pollen Allergy". ... "Effect of tree pollen specific, subcutaneous immunotherapy on the oral allergy syndrome to apple and hazelnut". Allergy . 59 (12): 1272–1276. doi : 10.1111/j.1398-9995.2004.00626.x .
A rare, genetic, renal disease characterized by the association of familial adult medullary cystic disease with spastic quadriparesis. There have been no further descriptions in the literature since 1990.
Clinical Features Verloes et al. (1990) described an Arabic sibship originating from Morocco in which 4 children had a syndrome of intrauterine growth retardation, microcephaly, large soft pinnae, telecanthus or true hypertelorism with squint, unusual hooked nose, narrow mouth, retrognathia, and severe neurologic impairment.
Resistance to thyrotropin-releasing hormone (TRH) syndrome is a type of central congenital hypothyroidism (see this term) characterized by low levels of thyroid hormones due to insufficient release of thyroid-stimulating hormone (TSH) caused by pituitary resistance to TRH.
A number sign (#) is used with this entry because of evidence that nongoitrous congenital hypothyroidism-7 (CHNG7) is caused by homozygous or compound heterozygous mutation in the TRHR gene (188545) on chromosome 8q23. Description Nongoitrous congenital hypothyroidism-7 (CHNG7) is characterized by normal-to-low T4 and normal-to-high thyrotropin (TSH; see 188540) levels, with reduced or absent pituitary responsiveness to thyrotropin-releasing hormone (TRH; 613879). Patients may exhibit short stature, growth retardation, and delayed bone age, as well as lethargy or fatigue (Collu et al., 1997; Bonomi et al., 2009). For a general phenotypic description and a discussion of genetic heterogeneity of congenital nongoitrous hypothyroidism, see 275200. Clinical Features Collu et al. (1997) studied a 12.5-year-old boy who presented at age 9 years with short stature.
The fetus had a normal 46,XX karyotype and a large nuchal bleb with no evidence of encephalocele. Nuchal bleb in the XO Turner syndrome is also accompanied by elevation of AFP in the amniotic fluid. ... In 11, the karyotype was consistent with the Turner syndrome and another probably had XY gonadal dysgenesis. ... Inheritance Although cystic hygroma is a conspicuous feature of the Turner syndrome and may occur in some mendelian disorders, its occurrence as an isolated autosomal recessive has been suggested by several reports (e.g., Watson et al., 1990 and Williams and Josephson, 1997). ... Misc - Fetal hydrops - Fetal death Neck - Nuchal bleb - Fetal cystic hygroma Lab - Normal karyotype - High amniotic fluid AFP Inheritance - Autosomal recessive - more often 45,X Turner syndrome ▲ Close
Cystic hygroma can be associated with a nuchal lymphangioma or a fetal hydrops . [5] Additionally, it can be associated with Down syndrome , Turner syndrome , [6] or Noonan syndrome . If it is diagnosed in the third trimester, then chances of association with Down syndrome are increased, but if diagnosed in the second trimester, then it is associated with Turner syndrome. ... Cystic hygromas are also often seen in Turner's syndrome, although a patient who does not have the syndrome can present with this condition. ... (March 2008). "Prenatally Diagnosed Turner Syndrome and Cystic Hygroma: Incidence and Reasons for Referrals" . ... CS1 maint: archived copy as title ( link ) ^ Bruno Bissonnette (2006-08-10). Syndromes: Rapid Recognition and Perioperative Implications .
Diagnostic methods Biologically, autoimmune lymphoproliferative syndrome with recurrent viral infections is characterized by slightly elevated double-negative T cells (DNTs), and defective Fas-mediated apoptosis of B, T, and NK lymphocytes.
Description Caspase 8 deficiency is a syndrome of lymphadenopathy and splenomegaly, marginal elevation of 'double-negative T cells' (DNT; T-cell receptor alpha/beta+, CD4-/CD8-), defective FAS-induced apoptosis, and defective T-, B-, and natural killer (NK)-cell activation, with recurrent bacterial and viral infections (summary by Madkaikar et al., 2011). ... Nomenclature Puck and Straus (2004) referred to caspase 8 deficiency as autoimmune lymphoproliferative syndrome type IIB; see 601859. In review articles, Teachey et al. (2009) stated that caspase 8 deficiency is distinct from ALPS and Madkaikar et al. (2011) stated that caspase 8 deficiency is an 'ALPS-related' disorder.
A number sign (#) is used with this entry because of evidence that Loeys-Dietz syndrome-5 (LDS5) is caused by heterozygous mutation in the TGFB3 gene (190230) on chromosome 14q24. Description Loeys-Dietz syndrome-5 (LDS5), also known as Rienhoff (pronounced REENhoff) syndrome, is characterized by syndromic presentation of aortic aneurysms involving the thoracic and/or abdominal aorta, with risk of dissection and rupture. ... Bertoli-Avella et al. (2015) summarized the clinical features in 10 families segregating heterozygous mutations in the TGFB3 gene, in which affected individuals exhibited syndromic aortic aneurysmal disease that showed significant overlap with Loeys-Dietz syndrome (LDS), although striking intrafamilial and interfamilial clinical variability was observed. ... Microscopic examination of dissected aortic wall from 1 patient showed elastic fiber fragmentation with increased collagen and proteoglycan deposition, reminiscent of findings in both Marfan syndrome (154700) and LDS. In pathology reports from 2 other families, only mild elastic fiber fragmentation was noted. ... Molecular Genetics In a 9-year-old girl with low muscle mass, growth retardation, and distal arthrogryposis, who also exhibited features of Marfan, Loeys-Dietz, and Beals (121050) syndromes but did not meet the established diagnostic criteria for those syndromes, Rienhoff et al. (2013) analyzed 6 genes known to be associated with those disorders, including TGFB2 (190220), TGFBR1 (190181), TGFBR2 (190182), SMAD3 (603109), FBN1 (134797), and FBN2 (612570), but found no mutations.
SVAS is a frequent feature of Williams-Beuren syndrome (WBS; 194050), a contiguous gene deletion syndrome that includes hemizygous deletion of the ELN gene. ... It also was said not to be associated with the facial manifestations of Williams syndrome, but the photographs seem to belie that conclusion: the configuration of the mouth in patients III-16 and III-18 who had SVAS documented by echocardiogram is very suggestive of the Williams syndrome and is quite different from that in their brother, III-17, who had a normal echocardiogram. Schmidt et al. (1989) concluded that isolated SVAS and Williams syndrome represent 'clinically distinct entities.' ... None had manifestations of Williams syndrome. O'Connor et al. (1985) studied 6 patients with supravalvular aortic stenosis; 3 had Williams syndrome, 2 had familial SVAS (presumably without evidence of Williams syndrome), and 1 had sporadic SVAS. ... Ewart et al. (1993) found that deletion involving 7q11.23 and resulting in hemizygosity of the elastin gene is responsible for the Williams-Beuren syndrome (194050). Deletions limited to the elastin gene appear to result in SVAS, whereas deletions spanning at least 114 kb lead to Williams-Beuren syndrome.
Supravalvular aortic stenosis (SVAS) is a heart defect that develops before birth. This defect is a narrowing (stenosis) of the large blood vessel that carries blood from the heart to the rest of the body (the aorta). The condition is described as supravalvular because the section of the aorta that is narrowed is located just above the valve that connects the aorta with the heart (the aortic valve). Some people with SVAS also have defects in other blood vessels, most commonly stenosis of the artery from the heart to the lungs (the pulmonary artery ). An abnormal heart sound during a heartbeat (heart murmur) can often be heard during a chest exam.
Differential diagnosis SVAS can be part of the Williams-Beuren syndrome caused by microdeletion of the 7q11-q23 region, including the elastin and many contiguous genes. SVAS associated with Williams-Beuren syndrome is identical to isolated SVAS, however Williams-Beuren syndrome is also associated with a characteristic face, behavioral disorders and hypercalcemia. ... Genetic counseling Except for the cases with Williams-Beuren syndrome, the disease is either sporadic or familial.
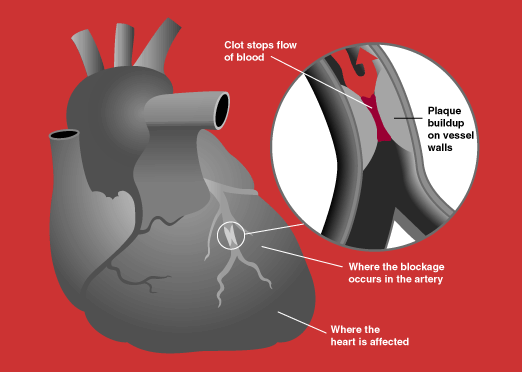
Overview Acute coronary syndrome is a term that describes a range of conditions related to sudden, reduced blood flow to the heart. ... Symptoms The symptoms of acute coronary syndrome usually begin suddenly. They include: Chest pain or discomfort. ... Risk factors The risk factors for acute coronary syndrome are the same as those for other types of heart disease. ... COVID-19 infection. Diagnosis Acute coronary syndrome requires emergency medical care at a hospital. ... Treatment The immediate goals of treatment for acute coronary syndrome are to: Relieve pain and distress.
Acute coronary syndrome Blockage of a coronary artery Specialty Cardiology Acute coronary syndrome ( ACS ) is a syndrome (set of signs and symptoms ) due to decreased blood flow in the coronary arteries such that part of the heart muscle is unable to function properly or dies . [1] The most common symptom is chest pain , often radiating to the left shoulder [2] or angle of the jaw, crushing, central and associated with nausea and sweating . ... This particular study had an 8.4% prevalence of acute coronary syndrome, which means the positive predictive value of being a male with chest pain and having coronary syndrome is 9.6% and negative predictive value is 93.2% ( click here to adjust these results for patients at higher or lower risk of acute coronary syndrome). ... See also [ edit ] Allergic acute coronary syndrome (Kounis syndrome) References [ edit ] ^ a b Amsterdam, E. ... "Clinical diagnosis of acute coronary syndrome in patients with chest pain and a normal or non-diagnostic electrocardiogram" . ... "Immediate vs delayed intervention for acute coronary syndromes: a randomized clinical trial" .
A rare hair disorder characterized by the appearance of lustreless, curly, frizzy, and coarse hair generally during adolescence predominantly in the frontal, temporal, and vertex regions of the scalp. Eyelashes, as well as growth and pigmentation of the hair, may also be affected.
A rare, acquired motor neuron disease characterized by a slowly progressive, unilateral, ascending or descending hemplegia, associated to unilateral or asymmetrical pyramidal signs and no sensory loss. It is a diagnosis of exclusion and contorversy exists regarding whether the presence of bulbar symptoms, sphincter disturbances, fasciculations or cognitive manifestations characterize the disease.