A rare genetic neurological disorder characterized by childhood-onset dystonia with distinctive MRI changes in the basal ganglia, and optic atrophy developing either immediately or within a few years after the appearance of dystonia. Additional symptoms include chorea and other movement disorders, dysarthria, or nystagmus, among others. Motor disability progresses gradually, while cognitive function is relatively spared.
A number sign (#) is used with this entry because of evidence that childhood-onset dystonia with optic atrophy and basal ganglia abnormalities (DYTOABG) is caused by homozygous or compound heterozygous mutation in the MECR gene (608205) on chromosome 1p35. Description Childhood-onset dystonia with optic atrophy and basal ganglia abnormalities is an autosomal recessive neurologic disorder characterized by onset of involuntary movements in the first decade of life. Optic atrophy develops around the same time or slightly later. Severity is variable, and some patients lose independent ambulation. Brain imaging shows abnormalities in the basal ganglia. Cognition appears to be unaffected (summary by Heimer et al., 2016). Clinical Features Heimer et al. (2016) reported 7 patients from 5 unrelated families with onset of involuntary movements, mainly dystonia, between 15 months and 6.5 years of age.
A rare genetic neurological disorder characterized by sensorineural hearing loss, sensory neuropathy, behavioral abnormalities, and dementia. Occurrence of seizures has also been reported. Age of onset is between adolescence and adulthood. The disease is progressive, with fatal outcome typically in the fifth to sixth decade.
A number sign (#) is used with this entry because hereditary sensory neuropathy type IE (HSN1E) is caused by heterozygous mutation in the DNMT1 gene (126375) on chromosome 19p13. Description Hereditary sensory neuropathy type IE is an autosomal dominant neurodegenerative disorder characterized by adult onset of progressive peripheral sensory loss associated with progressive hearing impairment and early-onset dementia (summary by Klein et al., 2011). For a discussion of genetic heterogeneity of HSN, see HSAN1A (162400). Clinical Features Wright and Dyck (1995) reported a 7-generation kindred with autosomal dominant inheritance of sensory neuropathy with sensorineural hearing loss and early-onset dementia. The neurologic deficits began between the second and fourth decades and were progressive, with death occurring in the fifth and sixth decades.
Hereditary sensory and autonomic neuropathy type IE (HSAN IE) is a disorder that affects the nervous system. It is characterized by three main features: hearing loss, a decline of intellectual function (dementia), and a worsening loss of sensation in the feet and legs (peripheral neuropathy). People with HSAN IE develop hearing loss that is caused by abnormalities in the inner ear (sensorineural hearing loss). The hearing loss, which affects both ears, gets worse over time and usually progresses to moderate or severe deafness between the ages of 20 and 35. Affected individuals experience dementia typically beginning in their thirties.
Hereditary sensory and autonomic neuropathy type 1E (HSAN1E) is a progressive disorder of the central and peripheral nervous systems . Symptoms typically begin by age 20 to 35 and include sensory impairment of the lower legs and feet; loss of sweating in the hands and feet; sensorineural hearing loss; and gradual decline of mental ability (dementia). The severity of symptoms and age of onset vary, even within the same family. HSAN1E is caused by a mutation in the DNMT1 gene and is inherited in an autosomal dominant manner. There is no effective treatment, but management may include injury prevention, the use of hearing aids, and sedative or antipsychotic medications for symptoms of dementia.
A rare genetic disease characterized by progressive and severe sensorineural hearing loss with onset in the first decade of life, associated with mild thrombocytopenia, often with enlarged platelets. Most patients do not show significant bleeding tendency.
These types can include myoclonic or absence seizures. In Dravet syndrome, these seizures are difficult to control with medication, and they can worsen over time. ... Many people with Dravet syndrome have difficulty coordinating movements (ataxia) and intellectual disability. ... The most commonly associated gene is SCN1A . More than 80 percent of Dravet syndrome cases and about 10 percent of other GEFS+ cases are caused by changes in this gene. ... Many mutations that cause Dravet syndrome reduce the number of functional channels in each cell. ... Other forms of GEFS+ are usually inherited from an affected parent. Rarely, Dravet syndrome or other forms of GEFS+ are inherited from a parent with somatic mosaicism .
Juvenile myoclonic epilepsy is a condition characterized by recurrent seizures (epilepsy). This condition begins in childhood or adolescence, usually between ages 12 and 18, and lasts into adulthood. The most common type of seizure in people with this condition is myoclonic seizures, which cause rapid, uncontrolled muscle jerks. People with this condition may also have generalized tonic-clonic seizures (also known as grand mal seizures), which cause muscle rigidity, convulsions, and loss of consciousness. Sometimes, affected individuals have absence seizures, which cause loss of consciousness for a short period that appears as a staring spell.
A number sign (#) is used with this entry because of evidence that Loeys-Dietz syndrome-4 (LDS4) is caused by heterozygous mutation in the TGFB2 gene (190220) on chromosome 1q41. ... Although features overlapping those of Marfan syndrome (MFS; see 154700) were found in some individuals, they were insufficient in any individual to meet the diagnostic criteria for MFS. ... Affected members of the family presented some minor connective tissue disease signs such as joint laxity, scoliosis, flat feet, dolichocephaly, and high palate; however, none met standard diagnostic criteria for Marfan (154700), Ehlers-Danlos (see 130000), or Loeys-Dietz syndromes or any other described disorder of connective tissue. ... Noting the substantial clinical and mechanistic overlap of this disorder with Loeys-Dietz syndrome (see 609192), Lindsay et al. (2012) suggested that categorizing it within the LDS spectrum would facilitate diagnosis and disease management. Using genomewide linkage analysis followed by whole-exome sequencing in 2 unrelated families with an autosomal dominant thoracic aortic aneurysm syndrome, 1 from the US and 1 French, who were negative for mutation in known aneurysm-related genes, Boileau et al. (2012) identified a frameshift and a nonsense mutation in the TGFB2 gene (190220.0003 and 190220.0004) that segregated with disease in each family.
Peroxisomal disorder Basic structure of a peroxisome Specialty Medical genetics Peroxisomal disorders represent a class of medical conditions caused by defects in peroxisome functions. [1] This may be due to defects in single enzymes [2] important for peroxisome function or in peroxins , proteins encoded by PEX genes that are critical for normal peroxisome assembly and biogenesis. [3] Contents 1 Peroxisome biogenesis disorders 2 Enzyme and transporter defects 3 References 4 External links Peroxisome biogenesis disorders [ edit ] Peroxisome biogenesis disorders (PBDs) include the Zellweger syndrome spectrum (PBD-ZSD) and rhizomelic chondrodysplasia punctata type 1 (RCDP1). [4] [5] PBD-ZSD represents a continuum of disorders including infantile Refsum disease , neonatal adrenoleukodystrophy , and Zellweger syndrome . ... "Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum". PMID 20301621 . Cite journal requires |journal= ( help ) ^ Steinberg, S.; Chen, L.; Wei, L.; Moser, A.; Moser, H.; Cutting, G.; Braverman, N. (2004). "The PEX Gene Screen: molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum". Molecular Genetics and Metabolism . 83 (3): 252–263. doi : 10.1016/j.ymgme.2004.08.008 . ... "Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders" . ... External links [ edit ] Classification D ICD - 10 : E80.3 ICD - 9-CM : 277.86 MeSH : D018901 External resources eMedicine : neuro/309 GeneReviews/NCBI/NIH/UW entry on Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum Peroxisomal+disorders at the US National Library of Medicine Medical Subject Headings (MeSH) The Global Foundation for Peroxisomal Disorders v t e Genetic disorder , organelle: Peroxisomal disorders and lysosomal structural disorders Peroxisome biogenesis disorder Zellweger syndrome Neonatal adrenoleukodystrophy Infantile Refsum disease Adult Refsum disease-2 RCP 1 Enzyme-related Acatalasia RCP 2&3 Mevalonate kinase deficiency D-bifunctional protein deficiency Adult Refsum disease-1 Transporter-related X-linked adrenoleukodystrophy Lysosomal Danon disease See also: proteins , intermediates
For the genetically-related condition, see McCune-Albright Syndrome . Albright's hereditary osteodystrophy Albright's hereditary osteodystrophy has an autosomal dominant pattern of inheritance Specialty Endocrinology Symptoms Choroid plexus calcification, Full cheeks [1] Causes Gs alpha subunit deficiency [2] Diagnostic method CBC, Urine test [3] Treatment Phosphate binders, supplementary calcium [4] Albright's hereditary osteodystrophy is a form of osteodystrophy , [5] and is classified as the phenotype of pseudohypoparathyroidism type 1A; this is a condition in which the body does not respond to parathyroid hormone . [1] Contents 1 Signs and symptoms 2 Genetics 3 Mechanism 4 Diagnosis 5 Treatment 6 History 7 See also 8 References 9 Further reading 10 External links Signs and symptoms [ edit ] The disorder is characterized by the following: [1] Hypogonadism Brachydactyly syndrome Choroid plexus calcification Hypoplasia of dental enamel Full cheeks Hypocalcemic tetany Choroid plexus(bottom left) Individuals with Albright hereditary osteodystrophy exhibit short stature , characteristically shortened fourth and fifth metacarpals , rounded facies , and often mild intellectual deficiency. [6] Albright hereditary osteodystrophy is commonly known as pseudohypoparathyroidism because the kidney responds as if parathyroid hormone were absent. ... Phosphate binders, supplementary calcium and vitamin D will be used as required. [4] History [ edit ] The disorder bears the name of Fuller Albright , who characterized it in 1942. [10] He was also responsible for naming it "Sebright bantam syndrome," after the Sebright bantam chicken, which demonstrates an analogous hormone insensitivity. Much less commonly, the term Martin-Albright syndrome is used, this refers to Eric Martin. [11] See also [ edit ] Pseudopseudohypoparathyroidism References [ edit ] ^ a b c "Albright's hereditary osteodystrophy" . ... Pseudo-hypoparathyroidism-example of 'Seabright-Bantam syndrome'; report of three cases. Endocrinology, Baltimore, 1942, 30: 922-932. ^ D. ... External links [ edit ] Classification D ICD - 10 : E20.1 ICD - 9-CM : 275.49 OMIM : 103580 MeSH : C537045 DiseasesDB : 10835 Scholia has a topic profile for Albright's hereditary osteodystrophy . v t e Bone and joint disease Bone Inflammation endocrine : Osteitis fibrosa cystica Brown tumor infection : Osteomyelitis Sequestrum Involucrum Sesamoiditis Brodie abscess Periostitis Vertebral osteomyelitis Metabolic Bone density Osteoporosis Juvenile Osteopenia Osteomalacia Paget's disease of bone Hypophosphatasia Bone resorption Osteolysis Hajdu–Cheney syndrome Ainhum Gorham's disease Other Ischaemia Avascular necrosis Osteonecrosis of the jaw Complex regional pain syndrome Hypertrophic pulmonary osteoarthropathy Nonossifying fibroma Pseudarthrosis Stress fracture Fibrous dysplasia Monostotic Polyostotic Skeletal fluorosis bone cyst Aneurysmal bone cyst Hyperostosis Infantile cortical hyperostosis Osteosclerosis Melorheostosis Pycnodysostosis Joint Chondritis Relapsing polychondritis Other Tietze's syndrome Combined Osteochondritis Osteochondritis dissecans Child leg: hip Legg–Calvé–Perthes syndrome tibia Osgood–Schlatter disease Blount's disease foot Köhler disease Sever's disease spine Scheuermann's_disease arm: wrist Kienböck's disease elbow Panner disease
Pseudohypoparathyroidism should not be confused with polyostotic fibrous dysplasia (174800), to which Albright's name is also attached as 'McCune-Albright syndrome.' Clinical Features Albright et al. (1942) described 3 patients with short stature, round face, short neck, obesity, subcutaneous calcifications, and shortened metacarpals associated with hypocalcemia and hyperphosphatemia. ... History Pseudohypoparathyroidism was the first endocrine syndrome in humans to be recognized as a failure of end-organ response to a hormone. ... Albright hereditary osteodystrophy, manic depressive psychosis (309200), and 'X-linked' pterygium syndrome (312150) were cited as examples of diseases compatible with X-linked dominance but not worse in males. ... Hedeland et al. (1992) described a mother and daughter with classic features of pseudohypoparathyroidism type Ia in association with proximal deletion of 15q, del(15)(q11q13), similar to that seen in Prader-Willi syndrome (176270) and Angelman syndrome (105830).
Pseudohypoparathyroidism type 1A (PHP1a) is a type of pseudohypoparathyroidism (PHP; see this term) characterized by renal resistance to parathyroid hormone (PTH), resulting in hypocalcemia, hyperphosphatemia, and elevated PTH; resistance to other hormones including thydroid stimulating hormone (TSH), gonadotropins and growth-hormone-releasing hormone (GHRH); and a constellation of clinical features known as Albright hereditary osteodystrophy (AHO; see this term). Epidemiology Prevalence of PHP (1a and 1b) has been estimated at 1/295,000 in a Japanese study. The prevalence of PHP (1a, 1b and pseudopseudohypoparathyroidism (PPHP); see these terms) has been estimated at 1/150,000in Italy. Clinical description Onset of endocrine symptoms occurs during childhood, although cases with severe hypothyroidism at neonatal screening have been reported. Patients present with varying degrees of AHO features (including obesity).
A number sign (#) is used with this entry because of evidence that rhizomelic chondrodysplasia punctata type 5 (RCDP5) is caused by homozygous mutation in the PEX5 gene (600414) on chromosome 12p13. Description Rhizomelic chondrodysplasia punctata (RCDP) is a peroxisomal disorder characterized by disproportionately short stature primarily affecting the proximal parts of the extremities, a typical facial appearance including a broad nasal bridge, epicanthus, high-arched palate, dysplastic external ears, and micrognathia, congenital contractures, characteristic ocular involvement, dwarfism, and severe mental retardation with spasticity. Biochemically, plasmalogen synthesis and phytanic acid alpha-oxidation are defective. Most patients die in the first decade of life (summary by Wanders and Waterham, 2005). For a discussion of genetic heterogeneity of rhizomelic chondrodysplasia punctata, see 215100.
A number sign (#) is used with this entry because rhizomelic chondrodysplasia punctata type 2 (RCDP2) is caused by homozygous or compound heterozygous mutation in the DHAPAT gene (GNPAT; 602744), which encodes acyl-CoA:dihydroxyacetonephosphate acyltransferase, on chromosome 1q42. Description Rhizomelic chondrodysplasia punctata (RCDP) is a peroxisomal disorder characterized by disproportionately short stature primarily affecting the proximal parts of the extremities, a typical facial appearance including a broad nasal bridge, epicanthus, high-arched palate, dysplastic external ears, and micrognathia, congenital contractures, characteristic ocular involvement, dwarfism, and severe mental retardation with spasticity. Biochemically, plasmalogen synthesis and phytanic acid alpha-oxidation are defective. Most patients die in the first decade of life. RCDP1 (215100) is the most frequent form of RCDP (summary by Wanders and Waterham, 2005). Whereas RCDP1 is a peroxisomal biogenesis disorder (PBD), RCDP2 is classified as a single peroxisome enzyme deficiency (Waterham and Ebberink, 2012).
Differential diagnosis The principle differential diagnosis is Zellweger syndrome. It also includes Warfarin embryopathy, fatty acyl-CoA reductase 1 deficiency, and Chondrodysplasia punctata, tibia-metacarpal type.
., X-linked chondrodysplasia punctata (see 302960); the multiple forms of the Zellweger syndrome (see 214100); maternal ingestion of certain anticoagulants (dicoumarol or warfarin; 118650) in early pregnancy; and even occasionally trisomy 18 (Rosenfield et al., 1962). ... In Australia this feature led to the designation koala bear syndrome (Danks, 1970). It was the suggestion of a group convened in Paris by the European Society of Pediatric Radiology that this phenotype be called chondrodysplasia punctata (Maroteaux, 1970). ... Because of clinical similarities to Zellweger syndrome, they did studies that showed evidence for their proposal. ... Wanders et al. (1986) did cell-fusion studies of complementation between RCDP and either Zellweger syndrome or the infantile form of Refsum disease (266500). ... Sheffield et al. (1989) concluded that Binder syndrome (155050) should be classified as a mild form of chondrodysplasia punctata.
Rhizomelic chondrodysplasia punctata (RCDP) is a type of peroxisomal disorder which impairs the normal development of many parts of the body. It is characterized by shortening of the bones in the upper arms and thighs (rhizomelia). People with RCDP have very poor growth and often develop joint deformities (contractures) that make the joints stiff and painful. Other major features include distinctive facial features, intellectual disability, clouding of the lenses of the eyes (cataracts), heart defects, and respiratory problems. There are 5 types of RCDP, classified according to the associated gene mutations: RCDP1 with PEX7 gene RCDP2 with GNPAT gene RCDP3 with AGPS gene RCDP4 (peroxisomal fatty acyl-CoA reductase 1 disorder) with FAR1 gene RCDP5 with PEX5 gene All these genes are involved in the formation and function of sac-like cell structures called peroxisomes that contain enzymes needed to break down many substances, including fatty acids known as plasmalogens.
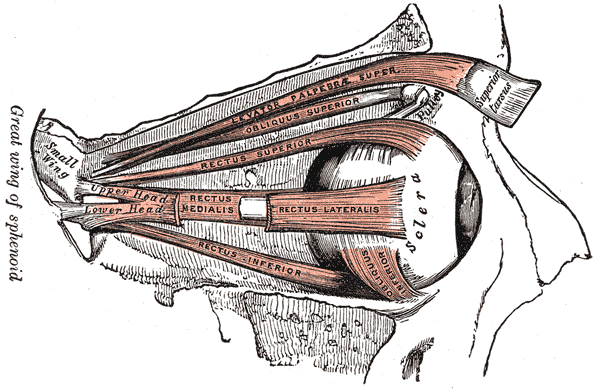
Phantom eye syndrome Anatomy of the eye . The external eye muscles are shown in red. Specialty Psychiatry , Neurology Duration 11-15 days Frequency 5% The phantom eye syndrome ( PES ) is a phantom pain in the eye and visual hallucinations after the removal of an eye ( enucleation , evisceration ). ... In contrast, visual hallucinations caused by severe visual loss without removal of the eye itself ( Charles Bonnet syndrome ) are less frequent (prevalence 10%) and often consist of detailed images. ... Treatment [ edit ] Treatment of painful phantom eye syndrome is provision of ocular prosthesis in the empty orbit. [2] See also [ edit ] Visual system Charles Bonnet syndrome Phantom limb References [ edit ] ^ a b c Sörös, P.; O. ... Gerding (May 2003). "Phantom eye syndrome: Its prevalence, phenomenology, and putative mechanisms".
Gonadoblastoma has been found in association with androgen insensitivity syndrome , mixed gonadal dysgenesis and Turner syndrome , especially in the presence of Y chromosome-bearing cells. [3] [4] Women with Turner syndrome whose karyotype includes a Y chromosome (as in 45,X/46,XY mosaicism) are at increased risk for gonadoblastoma. Because of the risk of gonadoblastoma, individuals with Turner syndrome with detectable Y chromosome material (Mosaic Turner syndrome) should have their gonads prophylactically removed. In a population-based study, the cumulative risk for women with Turner syndrome and Y chromosome material was 7.9 percent by age 25 years. [5] Diagnosis [ edit ] Classification [ edit ] Main article: Germ cell tumor Gonadoblastomas can contain elements of both germ cells and gonadal stroma. [6] Formerly, gonadoblastoma was sometimes regarded as a subset of dysgerminoma . ... "Polycystic ovary and gonadoblastoma in Turner's syndrome". Minerva Pediatr . 59 (4): 397–401. ... "Detection of hidden Y mosaicism in Turner's syndrome: importance in the prevention of gonadoblastoma".
By PCR, Gravholt et al. (2000) examined 114 females with Turner syndrome for the presence of Y-chromosomal material and found 14 who had Y-chromosomal material. ... The authors concluded that the frequency of Y-chromosomal material is high in Turner syndrome (12.2%), but the occurrence of gonadoblastoma among Y-positive patients seems to be low (7-10%), and the risk may have been overestimated in previous studies, perhaps due to problems with selection bias.
Gonadoblastoma is a rare benign neoplasm of mixed sex cord and germ cells, arising mostly in the dysgenic gonads of young women with a chromosome Y anomaly, presenting with abdominal enlargement, variable feminization or virilization or, in some cases, being asymptomatic. It is often associated with dysgerminoma.
A number sign (#) is used with this entry because of evidence that proteasome-associated autoinflammatory syndrome-3 (PRAAS3) is caused by homozygous mutation in the PSMB4 gene (602177) on chromosome 1q21. ... Description Proteasome-associated autoinflammatory syndrome-3 is an autosomal recessive syndrome with onset in early infancy. ... He was also noted to have myositis, joint contractures, hypergammaglobulinemia, antinuclear autoantibodies, thrombocytopenia, lymphopenia, and metabolic syndrome. Digenic Inheritance Brehm et al. (2015) reported 2 sibs of Jamaican descent (family 4) with digenic PRAAS (see MOLECULAR GENETICS). ... More variable features included metabolic syndrome and peripheral calcinosis. Inheritance The transmission pattern of PRAAS3 in a family (family 1) reported by Brehm et al. (2015) was consistent with autosomal recessive inheritance. ... INHERITANCE - Autosomal recessive GROWTH Other - Failure to thrive - Poor overall growth HEAD & NECK Head - Sinusitis Face - Facial edema Ears - Otitis Eyes - Periorbital swelling - Periorbital erythema - Violaceous eyelids - Conjunctivitis ABDOMEN Liver - Hepatomegaly Spleen - Splenomegaly SKELETAL - Arthralgia - Arthritis - Joint contractures Hands - Finger swelling Feet - Toe swelling SKIN, NAILS, & HAIR Skin - Skin rash - Skin lesions - Annular plaques - Acanthosis nigricans - Neutrophilic dermatosis MUSCLE, SOFT TISSUES - Lipodystrophy - Intraabdominal fat - Myositis - Panniculitis - Peripheral calcinosis NEUROLOGIC Central Nervous System - Aseptic meningitis METABOLIC FEATURES - Fever, recurrent - Metabolic syndrome HEMATOLOGY - Anemia - Thrombocytopenia - Lymphopenia IMMUNOLOGY - Hypergammaglobulinemia - Autoantibodies - Autoinflammation, chronic - Lymphadenopathy - Lymphopenia - Recurrent infections LABORATORY ABNORMALITIES - Increased acute phase reactants - Increased erythrocyte sedimentation rate - Increased C-reactive protein - Increased triglycerides - High LDL - Low HDL - Elevated liver enzymes MISCELLANEOUS - Onset in early infancy - One patient with only PSMB4 mutations has been reported (last curated July 2018) - Digenic inheritance (mutation in PSMB4 and PSMB9) has been reported in 1 family (last curated July 2018) MOLECULAR BASIS - Caused by mutation in the proteasome subunit, beta-type, 4 gene (PSMB4, 602177.0001 ) ▲ Close
Severe fever with thrombocytopenia syndrome SFTS bunyavirus isolated from patients in Central and Northeast provinces of China (red) Specialty Infectious disease Severe fever with thrombocytopenia syndrome ( SFTS ) is an emerging infectious disease caused by Dabie bandavirus also known as the SFTS virus , first reported between late March and mid-July 2009 in rural areas of Hubei and Henan provinces in Central China. [1] SFTS has fatality rates ranging from 12% to as high as 30% in some areas. ... "Person-to-person transmission of severe fever with thrombocytopenia syndrome virus" . Vector Borne and Zoonotic Diseases (Larchmont, N.Y.) . 12 (2): 156–160. doi : 10.1089/vbz.2011.0758 . ... "Evolutionary and molecular analysis of the emergent severe fever with thrombocytopenia syndrome virus" . Epidemics . 5 (1): 1–10. doi : 10.1016/j.epidem.2012.09.002 . ... External links [ edit ] Classification D ICD - 10 : A93.8 ICD - 9-CM : 065.9 MeSH : D002044 v t e Tick-borne diseases and infestations Diseases Bacterial infections Rickettsiales Anaplasmosis Boutonneuse fever Ehrlichiosis ( Human granulocytic , Human monocytotropic , Human E. ewingii infection ) Scrub typhus Spotted fever rickettsiosis Pacific Coast tick fever American tick bite fever rickettsialpox Rocky Mountain spotted fever ) Spirochaete Baggio–Yoshinari syndrome Lyme disease Relapsing fever borreliosis Thiotrichales Tularemia Viral infections Bhanja virus Bourbon virus Colorado tick fever Crimean–Congo hemorrhagic fever Heartland bandavirus Kemerovo tickborne viral fever Kyasanur Forest disease Omsk hemorrhagic fever Powassan encephalitis Severe fever with thrombocytopenia syndrome Tete orthobunyavirus Tick-borne encephalitis Protozoan infections Babesiosis Other diseases Tick paralysis Alpha-gal allergy Southern tick-associated rash illness Infestations Tick infestation Species and bites Amblyomma Amblyomma americanum Amblyomma cajennense Amblyomma triguttatum Dermacentor Dermacentor andersoni Dermacentor variabilis Ixodes Ixodes cornuatus Ixodes holocyclus Ixodes pacificus Ixodes ricinus Ixodes scapularis Ornithodoros Ornithodoros gurneyi Ornithodoros hermsi Ornithodoros moubata Other Rhipicephalus sanguineus