-
Achoo Syndrome
Omim
Lerner (1991) took Hunter (1990) to task for referring to the photic sneeze reflex as a 'comic syndrome.' He cited reports by Beckman and Nordenson (1983), Forrester (1985), Morris (1987), and Lang and Howland (1987), in addition to those already cited here. ... History Duncan (1995) pointed out public awareness of the ACHOO syndrome is much more widespread than one might guess, to the point that it has entered into the popular wisdom conveyed to preschoolers. ... Lehvaslaiho (2003) found an early reference to the ACHOO syndrome by Aristotle (Barnes, 1984). In Book XXXIII, in a section entitled 'Problems concerning the nose,' Aristotle stated: 'Why is it that one sneezes more after one has looked at the sun?ZFHX3, ESRRG, LINC00364, DPH6-DT, LINC01798, HULC, LINC00461, LINC00971, BRWD3, EMX2OS, KIAA1755, SRRM4, PRR5L, PKNOX2, ENOX1, TRIM44, RBFOX1, AUTS2, CAMTA1, KLF12, NXPH4, ADAMTS13, NRG3, AKAP6, FHIT, PKN2-AS1

-
Spatial Visualization, Aptitude For
Omim
Garron (1970) pointed out that if spatial and numerical abilities are determined by an X-linked recessive gene, patients with Turner syndrome should show superior not inferior performance. ... The rationale for studying a single large kindred was the possibility of reducing genetic heterogeneity and increasing the power of linkage studies. Turner syndrome (monosomy X) is associated with a characteristic neurocognitive profile that includes impaired visuospatial/perceptual abilities. Ross et al. (2000) used a molecular approach to identify a critical region of the X chromosome for neurocognitive aspects of Turner syndrome. Partial deletions of Xp in 34 females were mapped by FISH or by loss of heterozygosity of polymorphic markers. Discriminant function analysis optimally identified the Turner syndrome-associated neurocognitive phenotype. ... Ross et al. (2000) concluded that the haploinsufficiency of a PAR1 gene or genes is the basis for susceptibility to the Turner syndrome neurocognitive phenotype. Females with nonmosaic deletions missing only distal Xp22.33, at a minimum, manifested the defined Turner syndrome-associated cognitive profile.
-
Paganini-Miozzo Syndrome
Omim
A number sign (#) is used with this entry because of evidence that Paganini-Miozzo syndrome (MRXSPM) is caused by hemizygous mutation in the HS6ST2 gene (300545) on chromosome Xq26. One such family has been reported. Description Paganini-Miozzo syndrome (MRXSPM) is a neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development, high myopia, and mild dysmorphic facial features (summary by Paganini et al., 2019) Clinical Features Paganini et al. (2019) reported 2 monozygotic Italian twin brothers with a syndromic neurodevelopmental disorder.
-
Pterygia, Mental Retardation, And Distinctive Craniofacial Features
Omim
In a systematic survey of the mentally retarded, Haspeslagh et al. (1985) identified 3 females, related as first or second cousins, with a seemingly unique syndrome. They were short of stature and had an unusual combination of craniofacial features: trigonocephaly, bulging forehead, flat face, and microretrognathia. ... Even though multiple pterygia occurred in only 1 index case, the authors suggested that this is a key feature of the full-blown MCA/MR (multiple congenital anomalies-mental retardation) syndrome present in the others in somewhat milder form. ... They emphasized that severe mental retardation appears to distinguish this syndrome from the other multiple pterygium syndromes. However, van Bever and Hennekam (1995) described an adult female with features suggesting the Haspeslagh syndrome but without severe mental retardation and pterygia. ... This report confirmed that small chromosomal aberrations, difficult to detect by standard cytogenetic techniques, may cause familial private syndromes. INHERITANCE - Autosomal dominant GROWTH Height - Short stature HEAD & NECK Head - Trigonocephaly - Bulging forehead Face - Flat face - Microretrognathia Ears - Dysplastic ears Eyes - Epicanthal folds Mouth - Small mouth CHEST Ribs Sternum Clavicles & Scapulae - Pectus excavatum Breasts - Hypoplastic nipples GENITOURINARY External Genitalia (Male) - Genital hypoplasia - Hypospadias External Genitalia (Female) - Genital hypoplasia SKELETAL Feet - Club feet SKIN, NAILS, & HAIR Skin - Pterygia, multiple NEUROLOGIC Central Nervous System - Mental retardation - Seizures - Hypoplastic corpus callosum - Abnormal gyration LABORATORY ABNORMALITIES - High resolution G- and T- banding karyotype shows reciprocal translocation of distal 9p-6q ▲ Close
-
Kowarski Syndrome
Omim
A number sign (#) is used with this entry because of evidence that Kowarski syndrome is caused by mutation in the growth hormone gene (GH1; 139250) on chromosome 17q. Mutation in the GH1 gene also causes several types of isolated growth hormone deficiency (IGHD); see 262400 for a summary. Description Kowarski syndrome, or short stature associated with bioinactive growth hormone, is characterized clinically by normal or slightly increased GH secretion, pathologically low IGF1 (147440) levels, and normal catch-up growth on GH replacement therapy (Besson et al., 2005). ... Besson et al. (2005) described a Serbian patient with Kowarski syndrome who was homozygous for a mutation in the GH1 gene that disrupted the first disulfide bridge in growth hormone (139250.0021).POU1F1, RNPC3, GHRHR, GHRH, GHSR, KISS1R, KISS1, CSHL1, NR5A1, GH1, TG, TACR3, GNRH1, GNRHR, HESX1, SOX3, LHX4, GLI2, PROP1, OTX2, LHX3, IGF1, GHR, IGHD, IGFBP3, SLC20A1, BTK, PHEX, PAX6, FBXO8, GPR101, HGH1, FBRS, ALB, SDS, SOAT1, SHBG, BRD2, PRL, MMUT, POMC

-
Angioma, Hereditary Neurocutaneous
Omim
His brother developed left hemiparesis at age 13 and died at age 19 after an unsuccessful attempt was made to resect a spinal angioma in the C6-T1 region (producing the Horner syndrome and the Brown-Sequard syndrome). ... None of the patients had retinal angiomas or telangiectases typical of Osler-Rendu-Weber syndrome. The involvement of the central nervous system resembled that in the Icelandic family reported by Kidd and Cumings (1947) but that family had no skin angiomas. ... Foo et al. (1980) reported the case of a 33-year-old man who developed cervical anterior cord syndrome from spontaneous bleeding of an arteriovenous malformation in the cervical epidural space. ... Although the evidence is not ironclad, this syndrome of hereditary neurocutaneous angioma is probably distinct from familial cavernous malformations of the CNS and retina (116860). GU - Episodic hematuria Inheritance - Autosomal dominant Neuro - Multiple dilated thin-walled cerebral vessels - Spinal angioma - Horner syndrome - Hemiparesis GI - Gastrointestinal bleeding Skin - Large irregular flat hemangiomas ▲ Close
-
Mitochondrial Dna Depletion Syndrome, Hepatocerebral Form Due To Dguok Deficiency
Orphanet
Epidemiology Prevalence of Mitochondrial DNA depletion syndrome, hepatocerebral form due to DGUOK deficiency (DGUOK-MDS) is unknown. However, more than 100 cases of MDS have been described, DGUOK deficiency being one of the most common causes of hepatocerebral form of mitochondrial DNA depletion syndromes. Clinical description In most cases, DGUOK-MDS presents as a multi-organ disease in the first week of life with hypoglycemia and lactic acidosis followed by development of hepatic and neuromuscular dysfunction within weeks of birth. ... Differential diagnosis Differential diagnosis includes the other hepatocerebral mitochondrial depletion syndromes, namely due to mutations in the POLG , MPV17 or TWNK genes.
-
Staphylococcal Toxic-Shock Syndrome
Orphanet
Staphylococcal toxic shock syndrome (staphylococcal TSS) is an acute disease mediated by the production of superantigenic toxins, characterized by high fever, skin rash followed by skin peeling, hypotension, vomiting, diarrhea and potentially leading to multisystem organ failure and caused by a Staphylococcus aureus bacterial infection. ... More serious manifestations include confusion, shock, renal and myocardial dysfunction and acute respiratory distress syndrome (ARDS; see this term). ARDS is the most common cause of death seen in staphylococcal TSS. ... Laboratory analysis, where staphylococcal superantigens (such as toxic shock syndrome toxin 1) are identified in urine or serum, can aid in confirming diagnosis.
-
X-Linked Intellectual Disability, Cabezas Type
Orphanet
An X-linked syndromic intellectual disability characterized by developmental delay, intellectual disability (ID) with severe speech impairment, and short stature. ... Epidemiology The prevalence of Cabezas syndrome (CS) is unknown. It is estimated that CS accounts for 3% of X-linked intellectual disability. ... Females are mainly asymptomatic but in rare cases can present learning disability, attention deficit disorder, or tremor. Etiology This syndrome is caused by pathogenic variants in CUL4B gene (Xq24), encoding a scaffold protein of the cullin 4B-RING ubiquitin ligase (E3) complex, which is crucial in the regulation of the degradation of cellular proteins. ... Differential diagnosis Differential diagnosis includes Börjeson-Forssman-Lehmann syndrome, Wilson-Turner syndrome and Smith-Fineman-Myers syndrome.
-
Epilepsy, Hearing Loss, And Mental Retardation Syndrome
Omim
A number sign (#) is used with this entry because of evidence that epilepsy, hearing loss, and mental retardation syndrome (EHLMRS) is caused by homozygous or compound heterozygous mutation in the SPATA5 gene (613940) on chromosome 4q28. Description Epilepsy, hearing loss, and mental retardation syndrome is an autosomal recessive disorder characterized by severe neurologic impairment including intellectual disability, intractable epilepsy, microcephaly, abnormal muscle tone, and sensorineural hearing loss. ... Molecular Genetics In 14 children from 10 families with epilepsy, hearing loss, and mental retardation syndrome, Tanaka et al. (2015) identified homozygous or compound heterozygous mutations in the SPATA5 gene (see, e.g., 613940.0001-613940.0005).
-
Asherman Syndrome
Orphanet
Clinical description The severity of intrauterine adhesions (IUA) in Asherman's syndrome can vary between complete obliteration of the cavity to minimal, marginal adhesions. ... When the adhesions are exclusively located in the lower uterine tract and functioning endometrium persists, this syndrome can cause severe pelvic pain and retrograde menstruation. ... Diagnostic methods A history of uterine trauma (typically curettage) associated with menstrual abnormalities/inability to conceive are suggestive of Asherman's syndrome. Hysteroscopy provides definitive diagnosis, and can characterize the site and extent of adhesions as well as assess the endometrium.
-
Mitochondrial Complex I Deficiency, Nuclear Type 2
Omim
Clinical Features Loeffen et al. (1998) reported a patient with mitochondrial complex I deficiency manifesting as Leigh syndrome (see 256000). The patient presented at age 5 weeks with poor feeding and episodes of apnea and cyanosis. ... Procaccio and Wallace (2004) reported a patient with late onset of mitochondrial complex I deficiency manifesting as Leigh syndrome. The patient first developed walking difficulties at age 7 years, which progressed to balance impairment, dysarthria, mild dystonic posture, and nystagmus. ... The unrelated patient had mitochondrial encephalopathy, hypertrophic cardiomyopathy, hypotonia, and respiratory insufficiency. respectively. Both sibs had Leigh syndrome, hypotonia, and lactic acidosis; one had seizures and the other had hypertrophic cardiomyopathy. Molecular Genetics In a patient with mitochondrial complex I deficiency manifesting as Leigh syndrome, Loeffen et al. (1998) identified compound heterozygous mutations in the NDUFS8 gene (P79L, 602141.0001 and R102H, 602141.0002). ... In a patient with late onset of mitochondrial complex I deficiency manifesting as Leigh syndrome, Procaccio and Wallace (2004) identified compound heterozygous mutations in the NDUFS8 gene (P85L, 602141.0003 and R138H, 602141.0004).
-
Wasting
Wikipedia
Further information: Malnutrition This article is about the medical syndrome. For similar terms with different meanings, see Waste (disambiguation) . In medicine, wasting , also known as wasting syndrome , refers to the process by which a debilitating disease causes muscle and fat tissue to "waste" away. ... Infections and conditions associated with wasting include tuberculosis , chronic diarrhea , AIDS , and superior mesenteric artery syndrome . The mechanism may involve cachectin – also called tumor necrosis factor, a macrophage -secreted cytokine . ... Percent of body weight lost (At Tufts, an unintentional loss of 6% or more in 6 months is regarded as wasting) Treatment and prevention [ edit ] Antiretrovirals and anabolic steroids have been used to treat HIV wasting syndrome. [2] Additionally, an increase in protein-rich foods such as peanut butter , eggs , and cheese can assist in controlling the loss of muscle mass. [3] See also [ edit ] Anorexia Atrophy Cachexia Superior mesenteric artery syndrome Weight loss References [ edit ] ^ United Nations Children’s Fund, World Health Organization, The World Bank. ... Harrelson, Deidre Leaver-Dunn, "Principles of Pharmacology for Athletic Trainers", SLACK Incorporated, 2005, ISBN 1-55642-594-5 , p. 330 ^ "HIV wasting syndrome - HIV/AIDS" . www.hiv.va.gov .
-
Ohvira
Wikipedia
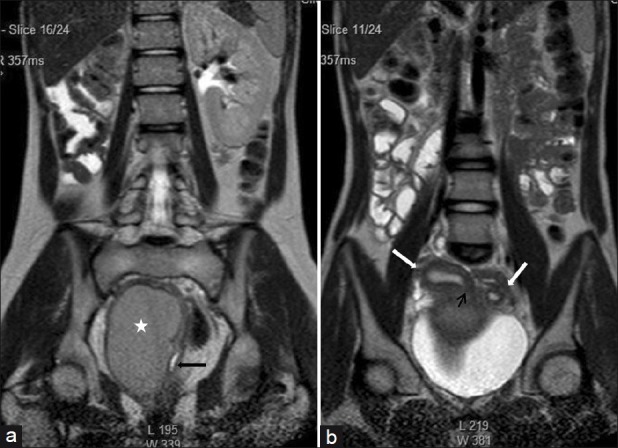
The right uterine horn cavity is seen to communicate with the upper end of the fluid collection in right hemivagina (small black arrow) Herlyn-Werner-Wunderlich syndrome , [1] also known as OHVIRA (Obstructed hemivagina and ipsilateral renal anomaly) is an extremely rare syndrome characterized by a congenital birth defect of the lower abdominal and pelvic organs. ... Pregnancies in women with Herlyn-Werner-Wünderlich syndrome are categorized as high-risk due to the size and shape of the uteri and cervices as well as the reduced kidney function. ... R.; Smitha, S. (2012-04-01). "OHVIRA syndrome (obstructed hemivagina and ipsilateral renal anomaly) with uterus didelphys, an unusual presentation". ... PMID 22421561 . ^ Aveiro, Ana Cristina; Miranda, Victor; Cabral, António Jorge; Nunes, Sidónia; Paulo, Filomeno; Freitas, Conceição (2011-07-15). "Herlyn–Werner–Wunderlich syndrome: a rare cause of pelvic pain in adolescent girls" . ... PMID 22689557 . ^ Tug, Niyazi; Sargin, Mehmet Akif; Çelik, Ayhan; Alp, Turgut; Yenidede, Ilter (2017-01-07). "Treatment of Virgin OHVIRA Syndrome with Haematometrocolpos by Complete Incision of Vaginal Septum without Hymenotomy" .
-
Dauwerse-Peters Syndrome
Omim
Cytogenetics By chromosome analysis in a man with this apparently novel brachydactyly-syndactyly syndrome, Dauwerse et al. (2007) identified a de novo translocation t(4;6)(q12;p23).
-
Trisomy 18-Like Syndrome
Omim
Shashi et al. (1996) concluded that the apparent mosaicism for trisomy 18 in the liver may have been spurious, and that the pattern of anomalies, together with the parental consanguinity, may indicate a new autosomal recessive malformation syndrome. The illustrated appearance of the patient was unusual with broad nasal root, narrow palpebral fissures, telecanthus, deficient alae nasi, apparently low-set ears which were malformed, preauricular tags, and micrognathia.
-
Craniosynostosis, Adelaide Type
Omim
Clinical Features Ades et al. (1994) described a southern Australian kindred with a form of craniosynostosis initially thought to represent the Jackson-Weiss syndrome (123150). They found in the hands coned epiphyses, distal and middle phalangeal hypoplasia, and carpal bone malsegmentation and in the feet coned epiphyses, hallux valgus, phalangeal, tarsonavicular and calcaneonavicular fusions. Mapping By linkage mapping in the family described by Ades et al. (1994), Hollway et al. (1995) excluded allelism to Saethre-Chotzen syndrome (101400) at 7p21, craniosynostosis type 2 (CRS2; 604757) at 5q34-q35, Jackson-Weiss and Crouzon (CFD1; 123500) syndromes at 10q24-q25, and Pfeiffer syndrome (101600) mapping near 8cen.
-
Armfield X-Linked Mental Retardation Syndrome
Omim
Mapping Linkage studies performed by Armfield et al. (1999) localized this apparently novel syndromic form of X-linked mental retardation to the terminal 8 Mb of Xq28, with a lod score of 2.11 at zero recombination at marker p39.
-
Craniofacial Anomalies And Anterior Segment Dysgenesis Syndrome
Omim
A number sign (#) is used with this entry because of evidence that craniofacial anomalies and anterior segment dysgenesis syndrome is caused by heterozygous mutation in the VSX1 gene (605020) on chromosome 20p11.
-
Inborn Errors Of Purine–pyrimidine Metabolism
Wikipedia
Inborn errors of purine–pyrimidine metabolism Specialty Endocrinology Inborn errors of purine–pyrimidine metabolism are a class of inborn error of metabolism disorders specifically affecting purine metabolism and pyrimidine metabolism . An example is Lesch–Nyhan syndrome . Urine tests may be of use in identifying some of these disorders. [1] References [ edit ] ^ Wevers RA, Engelke UF, Moolenaar SH, et al. ... External links [ edit ] Classification D ICD - 10 : E79 ICD - 9-CM : 277.2 MeSH : D011686 v t e Inborn error of purine–pyrimidine metabolism Purine metabolism Anabolism Adenylosuccinate lyase deficiency Adenosine Monophosphate Deaminase Deficiency type 1 Nucleotide salvage Lesch–Nyhan syndrome / Hyperuricemia Adenine phosphoribosyltransferase deficiency Catabolism Adenosine deaminase deficiency Purine nucleoside phosphorylase deficiency Xanthinuria Gout Mitochondrial neurogastrointestinal encephalopathy syndrome Pyrimidine metabolism Anabolism Orotic aciduria Miller syndrome Catabolism Dihydropyrimidine dehydrogenase deficiency This article about an endocrine, nutritional, or metabolic disease is a stub .


