-
Clouston's Hidrotic Ectodermal Dysplasia
Wikipedia
Find sources: "Clouston's hidrotic ectodermal dysplasia" – news · newspapers · books · scholar · JSTOR ( September 2015 ) ( Learn how and when to remove this template message ) Clouston's hidrotic ectodermal dysplasia Other names Alopecia congenita with keratosis palmoplantaris, Clouston syndrome, [1] : 571 Fischer–Jacobsen–Clouston syndrome, Hidrotic ectodermal dysplasia, Keratosis palmaris with drumstick fingers, Palmoplantar keratoderma and clubbing Specialty Dermatology , medical genetics Clouston's hidrotic ectodermal dysplasia is caused by mutations in a connexin gene, GJB6 or connexin-30, characterized by scalp hair that is wiry, brittle, and pale, often associated with patchy alopecia . [2] : 507,511,517–16 Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Screening 5 Treatment 6 See also 7 References 8 Further reading Presentation [ edit ] Hidrotic ectodermal dysplasia 2, or Clouston syndrome (referred to as HED2 throughout this entry) is characterized by partial or total alopecia, dystrophy of the nails, hyperpigmentation of the skin (especially over the joints), and clubbing of the fingers. ... See also [ edit ] Palmoplantar keratoderma List of cutaneous conditions List of cutaneous neoplasms associated with systemic syndromes References [ edit ] ^ James, William; Berger, Timothy; Elston, Dirk (2005). ... Further reading [ edit ] GeneReviews/NCBI/NIH/UW entry on Hidrotic Ectodermal Dysplasia 2 OMIM entries on Hidrotic Ectodermal Dysplasia 2 Classification D OMIM : 129500 v t e Diseases of ion channels Calcium channel Voltage-gated CACNA1A Familial hemiplegic migraine 1 Episodic ataxia 2 Spinocerebellar ataxia type-6 CACNA1C Timothy syndrome Brugada syndrome 3 Long QT syndrome 8 CACNA1F Ocular albinism 2 CSNB2A CACNA1S Hypokalemic periodic paralysis 1 Thyrotoxic periodic paralysis 1 CACNB2 Brugada syndrome 4 Ligand gated RYR1 Malignant hyperthermia Central core disease RYR2 CPVT1 ARVD2 Sodium channel Voltage-gated SCN1A Familial hemiplegic migraine 3 GEFS+ 2 Febrile seizure 3A SCN1B Brugada syndrome 6 GEFS+ 1 SCN4A Hypokalemic periodic paralysis 2 Hyperkalemic periodic paralysis Paramyotonia congenita Potassium-aggravated myotonia SCN4B Long QT syndrome 10 SCN5A Brugada syndrome 1 Long QT syndrome 3 SCN9A Erythromelalgia Febrile seizure 3B Paroxysmal extreme pain disorder Congenital insensitivity to pain Constitutively active SCNN1B / SCNN1G Liddle's syndrome SCNN1A / SCNN1B / SCNN1G Pseudohypoaldosteronism 1AR Potassium channel Voltage-gated KCNA1 Episodic ataxia 1 KCNA5 Familial atrial fibrillation 7 KCNC3 Spinocerebellar ataxia type-13 KCNE1 Jervell and Lange-Nielsen syndrome Long QT syndrome 5 KCNE2 Long QT syndrome 6 KCNE3 Brugada syndrome 5 KCNH2 Short QT syndrome KCNQ1 Jervell and Lange-Nielsen syndrome Romano–Ward syndrome Short QT syndrome Long QT syndrome 1 Familial atrial fibrillation 3 KCNQ2 BFNS1 Inward-rectifier KCNJ1 Bartter syndrome 2 KCNJ2 Andersen–Tawil syndrome Long QT syndrome 7 Short QT syndrome KCNJ11 TNDM3 KCNJ18 Thyrotoxic periodic paralysis 2 Chloride channel CFTR Cystic fibrosis Congenital absence of the vas deferens CLCN1 Thomsen disease Myotonia congenita CLCN5 Dent's disease CLCN7 Osteopetrosis A2, B4 BEST1 Vitelliform macular dystrophy CLCNKB Bartter syndrome 3 TRP channel TRPC6 FSGS2 TRPML1 Mucolipidosis type IV Connexin GJA1 Oculodentodigital dysplasia Hallermann–Streiff syndrome Hypoplastic left heart syndrome GJB1 Charcot–Marie–Tooth disease X1 GJB2 Keratitis–ichthyosis–deafness syndrome Ichthyosis hystrix Bart–Pumphrey syndrome Vohwinkel syndrome ) GJB3 / GJB4 Erythrokeratodermia variabilis Progressive symmetric erythrokeratodermia GJB6 Clouston's hidrotic ectodermal dysplasia Porin AQP2 Nephrogenic diabetes insipidus 2 See also: ion channels
-
Ciliopathy
Wikipedia
One emerging aspect is the wide spectrum of ciliopathy gene mutations found within different diseases." [8] List of ciliopathies [ edit ] "The phenotypic parameters that define a ciliopathy may be used to both recognize the cellular basis of a number of genetic disorders and to facilitate the diagnosis and treatment of some diseases of unknown" cause. [6] Known ciliopathies [ edit ] Condition OMIM Gene(s) Systems/organs affected Alström syndrome [6] [1] 203800 ALMS1 Asphyxiating thoracic dysplasia (Jeune syndrome) [6] [15] 208500 Bardet–Biedl syndrome [6] [5] [8] 209900 BBS1 , BBS2 , ARL6 , BBS4 , BBS5 , MKKS , BBS7 , TTC8 , BBS9 , BBS10 , TRIM32 , BBS12 Ellis–van Creveld syndrome [15] 225500 EVC , EVC2 Joubert syndrome [6] [8] 213300 INPP5E , TMEM216 , AHI1 , NPHP1 , CEP290 , TMEM67 , RPGRIP1L , ARL13B , CC2D2A , BRCC3 Brain Leber congenital amaurosis [15] 204000 GUCY2D , RPE65 McKusick–Kaufman syndrome [15] 236700 MKKS Meckel–Gruber syndrome [6] [8] [16] 249000 MKS1 , TMEM67 , TMEM216 , CEP290 , RPGRIP1L , CC2D2A Liver, heart, bone Nephronophthisis [6] [5] [8] 256100 NPHP1 , INVS , NPHP3 , NPHP4 , IQCB1 , CEP290 , GLIS2 , RPGRIP1L Kidney Orofaciodigital syndrome 1 [1] [5] 311200 OFD1 Polycystic kidney disease [6] [5] ( ADPKD and ARPKD ) [17] 173900 PKD1 , PKD2 , PKHD1 Kidney Primary ciliary dyskinesia (Kartagener syndrome) [6] 244400 DNAI1 , DNAH5 , TXNDC3 , DNAH11 , DNAI2 , KTU , RSPH4A , RSPH9 , LRRC50 Senior–Løken syndrome [5] 266900 NPHP1 , NPHP4 , IQCB1 , CEP290 , SDCCAG8 Eye Sensenbrenner syndrome (cranioectodermal dysplasia) [15] 218330 IFT122 Short rib–polydactyly syndrome [15] 613091 DYNC2H1 ? ... IFT88 Novel form of congenital anosmia , reported in 2012 [18] Likely ciliopathies [ edit ] Condition OMIM Gene(s) Systems/organs affected Acrocallosal syndrome [15] 200990 KIF7 , GLI3 Acromelic frontonasal dysostosis [15] 603671 ZSWIM6 Arima syndrome [15] 243910 Biemond syndrome [15] 113400 COACH syndrome [15] 216360 TMEM67 , CC2D2A , RPGRIP1L Conorenal syndrome [19] [15] 266920 Greig cephalopolysyndactyly syndrome [15] 175700 GLI3 Hydrolethalus syndrome [15] 236680 HYLS1 Johanson–Blizzard syndrome [15] 243800 UBR1 Mohr syndrome ( oral-facial-digital syndrome type 2) [15] 252100 Neu–Laxova syndrome [15] 256520 PHGDH , PSAT1 , PSPH Opitz G/BBB syndrome [15] 300000 MID1 Pallister–Hall syndrome [15] 146510 GLI3 Papillorenal syndrome [15] 120330 PAX2 Renal–hepatic–pancreatic dysplasia [15] 208540 NPHP3 Varadi–Papp syndrome (oral-facial-digital syndrome type 6) [15] 277170 Possible ciliopathies [ edit ] Condition OMIM Gene(s) Systems/organs affected Acrofacial dysostosis [15] Acrofrontofacionasal dysostosis 2 [15] 239710 Adams–Oliver syndrome [15] 100300 ARHGAP31 , DOCK6 , RBPJ , EOGT , NOTCH1 , DLL4 Asplenia with cardiovascular anomalies (Ivemark syndrome) [15] 208530 Autosomal recessive spastic paraplegia [15] Barakat syndrome (HDR syndrome) [15] 146255 GATA3 Basal cell nevus syndrome [15] 109400 PTCH1 , PTCH2 , SUFU Branchio‐oculo‐facial syndrome [15] 113620 TFAP2A C syndrome (Opitz trigonocephaly) [15] 211750 CD96 Carpenter syndrome [15] 201000 RAB23 Cephaloskeletal dysplasia (microcephalic osteodysplastic primordial dwarfism type 1) [15] 210710 RNU4ATAC Cerebrofaciothoracic dysplasia [15] 213980 TMCO1 Cerebrofrontofacial syndrome (Baraitser–Winter syndrome) [15] 243310 ACTB Cerebrooculonasal syndrome [15] 605627 Autosomal recessive spastic ataxia of Charlevoix-Saguenay [15] 270550 SACS Chondrodysplasia punctata 2 [15] 302960 EBP Choroideremia [15] 303100 CHM Chudley–McCullough syndrome [15] 604213 GPSM2 C‐like syndrome [15] 605039 ASXL1 Coffin–Siris syndrome [15] 135900 ARID1B , SOX11 , ARID2 Cohen syndrome [15] 216550 VPS13B Craniofrontonasal dysplasia [15] 304110 EFNB1 Dysgnathia complex [15] 202650 Ectrodactyly–ectodermal dysplasia–cleft syndrome type 1 [15] 129900 Endocrine–cerebroosteodysplasia syndrome [15] 612651 ICK Focal dermal hypoplasia [15] 305600 PORCN Frontonasal dysplasia [15] 136760 ALX3 , ALX4 , ALX1 Fryns microphthalmia syndrome [15] 600776 Fryns syndrome [15] 229850 Genitopatellar syndrome [15] 606170 KAT6B Hemifacial microsomia [15] 164210 Hypothalamic hamartomas [15] 241800 Johnson neuroectodermal syndrome [15] 147770 Juvenile myoclonic epilepsy [20] 254770 Kabuki syndrome [15] 147920 KMT2D , KDM6A Kallmann syndrome [15] 308700 ANOS1 Lenz–Majewski hyperostotic dwarfism [15] 151050 PTDSS1 Lissencephaly 3 [15] 611603 TUBA1A Marden–Walker syndrome [6] [15] 248700 PIEZO2 MASA syndrome [15] 303350 L1CAM Microhydranencephaly [15] 605013 NDE1 Mowat–Wilson syndrome [15] 235730 ZEB2 NDH syndrome [15] 610199 GLIS3 Oculoauriculofrontonasal syndrome [15] 601452 Oculocerebrocutaneous syndrome [15] 164180 Oculodentodigital dysplasia [15] 164200 GJA1 Optiz–Kaveggia syndrome [15] 305450 MED12 Otopalatodigital syndrome 2 [15] 304120 FLNA Periventricular heterotopia X‐linked [15] 300049 FLNA Perlman syndrome [15] 267000 DIS3L2 Pitt–Hopkins syndrome [15] 610954 TCF4 Polycystic liver disease [6] 174050 Proteus syndrome [15] 176920 AKT1 Pseudotrisomy 13 [15] 264480 Retinal cone dystrophy 1 [15] 180020 Some forms of retinitis pigmentosa [6] [21] [15] 268000 Robinow syndrome [15] 268310 ROR2 Rubinstein–Taybi syndrome [15] 180849 CREBBP Sakoda complex [15] 610871 Schinzel–Giedion syndrome [15] 269150 SETBP1 Split-hand/foot malformation 3 [15] 246560 Spondyloepiphyseal dysplasia congenita [15] 183900 COL2A1 Thanatophoric dysplasia [15] 187600 FGFR3 Townes–Brocks syndrome [15] 107480 SALL1 , DACT1 Tuberous sclerosis [15] 191100 TSC1 , TSC2 VATER association [15] 192350 Ven den Ende–Gupta syndrome [15] 600920 SCARF2 Visceral heterotaxy [15] 606325 Walker–Warburg syndrome [15] 236670 Warburg Micro syndrome [15] 615663 RAB3GAP1 X‐linked congenital hydrocephalus [15] 307000 L1CAM X‐linked lissencephaly [15] 300067 DCX Young–Simpson syndrome [15] 603736 KAT6B History [ edit ] Although non-motile or primary cilia were first described in 1898, they were largely ignored by biologists. ... For example, in just a single area of human disease physiology, cystic renal disease , cilia-related genes and proteins have been identified to have causal effect in polycystic kidney disease , nephronophthisis , Senior–Løken syndrome type 5, orofaciodigital syndrome type 1 and Bardet–Biedl syndrome . [5] References [ edit ] ^ a b c d e f g Adams, M.; Smith, U. ... The Clinical, Molecular, and Functional Genetics of Bardet-Biedl Syndrome , in Genetics of Obesity Syndromes . ... External links [ edit ] Classification D MeSH : D002925 DiseasesDB : 29887 External resources Orphanet : 363250 The Ciliary Proteome Web Page at Johns Hopkins v t e Diseases of cilia Structural receptor: Polycystic kidney disease cargo: Asphyxiating thoracic dysplasia basal body : Bardet–Biedl syndrome mitotic spindle : Meckel syndrome centrosome : Joubert syndrome Signaling Nephronophthisis Other/ungrouped Alström syndrome Primary ciliary dyskinesia Senior–Løken syndrome Orofaciodigital syndrome 1 McKusick–Kaufman syndrome Autosomal recessive polycystic kidney See also: ciliary proteinsWDR19, OFD1, KIF7, CEP290, TMEM67, RPGRIP1L, BBS2, DYNC2H1, MKS1, AHI1, MKKS, ARL13B, CPLANE1, IQCB1, NEK1, NEK8, CC2D2A, RPGR, SDCCAG8, TTC21B, ZNF423, NPHP1, NPHP3, WDR35, CEP164, PKHD1, RPGRIP1, LCA5, IFT80, XPNPEP3, TULP1, EVC2, BBS5, GLIS2, CRB1, UMOD, TMEM231, TMEM216, CCDC28B, BBS9, TCTN2, INVS, B9D1, TRIM32, FAM92A, TMEM237, USH2A, BBS1, NPHP4, BBS4, GUCY2D, GDF1, PCARE, TOPORS, USH1C, NR1H4, CCDC40, DNAAF2, LRAT, B9D2, FOXH1, DNAH11, ADGRV1, ARL6, IMPDH1, CDH23, BBS10, EVC, CCDC39, WDPCP, DNAAF3, CRELD1, CFTR, CRX, NKX2-5, TMEM138, RD3, PCDH15, DNAH5, TCTN1, WHRN, ATXN10, AIPL1, DNAH8, ZIC3, RDH12, VHL, BBS12, SCNN1A, CLRN1, SPATA7, MYO7A, CEP41, IFT43, PKD2, SCNN1G, LRRC56, TTC8, DNAAF1, USH1G, NODAL, ACVR2B, SCNN1B, RPE65, LEFTY2, BBS7, KCNJ13, HYLS1, TSC2, TSC1, SCLT1, WDR11, NEK4, IFT140, INPP5E, KIAA0586, CEP120, IFT122, IFT52, ALMS1, MAK, TMEM107, IFT172, PRKD1, DCDC2, FGFR1OP, CEP104, STK11, WDR60, TCTN3, C2CD3, CILK1, POC1B, IFT27, TGFB1, RHO, ARMC9, CFAP410, CRB2, FOXJ1, CENPF, MGS, USP35, ARL13A, NPHP3-ACAD11, TTC26, MCIDAS, MICAL3, DNAAF4, PIFO, FOPNL, NEK9, IFT20, UCN2, CEP19, TAPT1, USP38, CPLANE2, TBC1D32, FAM161A, TEKT1, SLC41A1, BICC1, CSPP1, TXNDC15, FAM149B1, ACTB, NINL, ADAMTS9, PPT1, MCM2, RAB8A, NME3, NOTCH2, ORC1, PAFAH1B1, PAM, PCM1, PCNT, PCP4, PDE6D, PIK3CA, PIK3CB, PIK3CD, PIK3CG, KPNA3, KIF11, JBS, CTNNB1, AGXT, ATD, ATR, CCND1, BUB1B, CETN2, DAP, IGF1, E2F4, FGF8, FGFR3, GAS8, GLI1, GLI2, PKD1, RAF1, CEP72, RFX1, WDR5, ACVR1, RCOR1, ARL2BP, POC1A, CHTOP, TRAF3IP1, SGSM3, IFT81, PIK3R4, HSPB11, SUFU, RAB23, IFT57, CEP55, B4GAT1, B3GNT2, KIF14, IFT88, RFX3, RNASE3, CCL2, HNF1A, TNF, EZR, TRRAP, CEP350, IKBKG, CDK10, TNFSF14, UNC119, USP8, KIAA0753, LCA10

-
Arthrogryposis–renal Dysfunction–cholestasis Syndrome
Wikipedia
Arthrogryposis–renal dysfunction–cholestasis syndrome Other names ARC syndrome [1] Specialty Dermatology Arthrogryposis–renal dysfunction–cholestasis syndrome is a cutaneous condition caused by a mutation in the VPS33B gene. [2] Most of the cases have been survived for infancy. Recently, College of Medical Sciences in Nepal reports a case of ARC syndrome in a girl at the age of more than 18 years. ... "Orphanet: Arthrogryposis renal dysfunction cholestasis syndrome" . www.orpha.net . Retrieved 18 May 2019 . ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... External links [ edit ] Classification D ICD - 10 : Q89.7 OMIM : 208085 MeSH : C535382 External resources Orphanet : 2697 v t e Inherited disorders of trafficking / vesicular transport proteins Vesicle formation Lysosome / Melanosome : HPS1 – HPS7 Hermansky–Pudlak syndrome LYST Chédiak–Higashi syndrome COPII : SEC23A Cranio-lenticulo-sutural dysplasia COG7 CDOG IIE APC: AP1S2 X-linked intellectual disability AP3B1 Hermansky–Pudlak syndrome 2 AP4M1 CPSQ3 Rab ARL6 BBS3 RAB27A Griscelli syndrome 2 CHM Choroideremia MLPH Griscelli syndrome 3 Cytoskeleton Myosin : MYO5A Griscelli syndrome 1 Microtubule : SPG4 Hereditary spastic paraplegia 4 Kinesin : KIF5A Hereditary spastic paraplegia 10 Spectrin : SPTBN2 Spinocerebellar ataxia 5 Vesicle fusion Synaptic vesicle : SNAP29 CEDNIK syndrome STX11 Hemophagocytic lymphohistiocytosis 4 Caveolae : CAV1 Congenital generalized lipodystrophy 3 CAV3 Limb-girdle muscular dystrophy 2B , Long QT syndrome 9 Vacuolar protein sorting : VPS33B ARC syndrome VPS13B Cohen syndrome DYSF Distal muscular dystrophy See also vesicular transport proteins This dermatology article is a stub .
-
Mohr Syndrome
Omim
Haumont and Pelc (1983) described 2 sisters who, in addition to the usual features of the Mohr syndrome, had the Dandy-Walker syndrome. They suggested that the association of brain abnormalities may reflect the existence of a second distinct form of the Mohr syndrome. Egger and Baraitser (1984) suggested that the sibs reported by Gustavson et al. (1971) and by Haumont and Pelc (1983) had Joubert syndrome (213300), not Mohr syndrome. Goldstein and Medina (1974) described talon cusps in Mohr syndrome. Silengo et al. (1987) presented 2 patients who they suggested lent support to the idea that Mohr syndrome and Majewski syndrome (263520) are mild and severe expressions, respectively, of the same autosomal recessive disorder. The 2 patients had features typical of OFD II or Mohr syndrome but also had laryngeal anomalies and hallucal and postaxial polysyndactyly of the feet typical of Majewski syndrome. ... He suggested autosomal recessive inheritance, which seems to be supported by the observation by Gorlin (1967) of the same syndrome in 2 sisters. Nomenclature Rimoin and Edgerton (1967) suggested that Mohr syndrome might be called the oral-facial-digital syndrome II.
-
Nevus Flammeus Nuchae
Wikipedia
. ^ MedlinePlus Encyclopedia : Stork bite External links [ edit ] Classification D OMIM : 163100 v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark

-
Laryngo-Onycho-Cutaneous Syndrome
Wikipedia
Laryngoonychocutaneous syndrome Specialty Dermatology Laryngo-onycho-cutaneous syndrome (also known as Shabbir syndrome ) is a rare epithelial disorder inherited in an autosomal recessive fashion. [1] [2] It is characterized by abnormalities in the larynx , nails (" onycho- "), and skin ("cutaneous"). [3] The disorder is only found in Punjabi Muslims and only a few cases have been reported. [4] It was characterized by Pakistani dermotologist Syed Ghulam Shabbir (1923–2002) [5] in 1986. [6] [7] It may be associated with LAMA3 . [8] See also [ edit ] Watson syndrome List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... S2CID 221861310 . ^ "Laryngo-onycho-cutaneous syndrome: MedlinePlus Genetics" . medlineplus.gov . ... "An unusual N-terminal deletion of the laminin 3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome" . Human Molecular Genetics . 12 (18): 2395–2409. doi : 10.1093/hmg/ddg234 . ... "An unusual N-terminal deletion of the laminin alpha3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome" . Hum. Mol. Genet . 12 (18): 2395–409. doi : 10.1093/hmg/ddg234 . ... External links [ edit ] Laryngo-onycho-cutaneous syndrome on MedlinePlus Classification D OMIM : 245660 MeSH : C537032 v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins This dermatology article is a stub .
-
C Syndrome
Omim
Description The C syndrome, also known as Opitz trigonocephaly syndrome, is a malformation syndrome characterized by trigonocephaly, severe mental retardation, hypotonia, variable cardiac defects, redundant skin, and dysmorphic facial features, including upslanted palpebral fissures, epicanthal folds, depressed nasal bridge, and low-set, posteriorly rotated ears (summary by Kaname et al., 2007). C syndrome shows phenotypic overlap with Bohring-Opitz syndrome, or C-like syndrome (605039), a disorder with more severe features than C syndrome, caused by heterozygous mutation in the ASXL1 gene (612990) on chromosome 20q11. ... Haaf et al. (1991) described this syndrome in the daughter of consanguineous parents. ... Osaki et al. (2006) described a newborn infant who had many clinical features similar to those of the C-like syndrome but did not have exophthalmos, which had been regarded as a hallmark of the C-like syndrome. ... Kaname et al. (2007) noted that this patient had relatively severe features for C syndrome, but also stated that it was uncertain whether there is (1) a spectrum in the C syndrome from the mild form (C syndrome) to the severe form (C-like syndrome), or (2) genetic heterogeneity among the patients with the C syndrome.
-
Harlequin Syndrome
Gard
Harlequin syndrome is a syndrome affecting the autonomic nervous system . ... The symptoms associated with Harlequin syndrome may be more likely to occur when a person has been exercising, is very warm, or is in an intense emotional situation. ... The exact cause of Harlequin syndrome is not completely understood. In some patients with this syndrome, the underlying cause seems to be a lesion or tumor that is affecting the ability of the cells of the autonomic nervous system to communicate with one side of the body. However, in many cases an exact cause of the symptoms is not found. Diagnosis of Harlequin syndrome is based on observing symptoms consistent with the syndrome, followed by a series of tests to rule out other diseases associated with Harlequin sign. Treatment may consist of removing any lesion that may be causing the symptoms of the syndrome. If no lesion is present and the syndrome is not interfering with a person’s daily living, treatment may not be necessary.

-
Neuro-Cardio-Facial-Cutaneous Syndromes
Wikipedia
Neuro-cardio-facial-cutaneous-syndromes (NCFC), (also referred to as neuro-craniofacial-cardiac syndromes ) is a group of developmental disorders with a genetic ground, affecting the nervous system, circulatory system, (cranio)facial and cutaneous development. These four parts are the common denominator for the syndromes, but are mostly accompanied by disturbances in other parts of the body. The two most common syndromes under this "umbrella" are: Leopard syndrome (LS) and Noonan syndrome (NS). Other members are Costello syndrome (CS), cardio-facio-cutaneous syndrome (CFC) and Neurofibromatosis type I (NF1). ... Mutations within the ERK signal transduction pathway appears to be a common cause for NCFC syndromes. Some autistic children also have craniofacial and cardiac defects. [2] See also [ edit ] SPRED1 gene Legius syndrome References [ edit ] ^ http://www.jci.org/articles/view/34385 ^ https://www.sciencedaily.com/releases/2009/02/090209094403.htm
-
17q21.31 Microdeletion Syndrome
Wikipedia
Rare genetic disorder caused by a deletion of six genes 17q21.31 microdeletion syndrome (Koolen–de Vries syndrome) Other names Koolen–De Vries syndrome, Koolen de Vries syndrome, Koolen De Vries syndrome 17q21.31 microdeletion syndrome , also known as Koolen–de Vries syndrome ( KdVS ), is a rare genetic disorder caused by a deletion of a segment of chromosome 17 which contains six genes. ... A. de Vries, who helped discover the syndrome in 2006. [3] References [ edit ] ^ a b Sharp AJ, Hansen S, Selzer RR, et al. ... "A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism". ... "Cutaneous features in 17q21.31 deletion syndrome: a differential diagnosis for cardio-facio-cutaneous syndrome" . ... External links [ edit ] Classification D OMIM : 610443 External resources GeneReviews : 17q21.31 microdeletion syndrome Orphanet : 363958 DECIPHER database entry for 17q21.31 microdeletion syndrome Orphanet entry for 17q21.31 microdeletion syndrome v t e Mutation Mechanisms of mutation Insertion Deletion Substitution Transversion Transition Mutation with respect to structure Point mutation Nonsense mutation Missense mutation Conservative mutation Silent mutation Frameshift mutation Dynamic mutation Large-scale mutation Chromosomal translocations Chromosomal inversions Mutation with respect to overall fitness Deleterious mutation Advantageous mutation Neutral mutation Nearly neutral mutation Synonymous mutation Nonsynonymous mutation v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22
-
Brugada Syndrome
Mayo_clinic
Symptoms Brugada syndrome often doesn't cause any noticeable symptoms. ... If your parent, sibling or child has been diagnosed with Brugada syndrome, you may want to ask your provider if you should have testing to see if you're at risk of Brugada syndrome. ... Men are more frequently diagnosed with Brugada syndrome than are women. Race. Brugada syndrome occurs more frequently in Asians than in people of other races. Fever. A fever doesn't cause Brugada syndrome, but it can irritate the heart and trigger fainting or sudden cardiac arrest in people with Brugada syndrome, especially children. ... Brugada syndrome complications include: Sudden cardiac arrest.SCN5A, SCN10A, PKP2, CACNA2D1, KCNE5, CACNA1C, KCND3, SCN1B, RANGRF, TRPM4, SCN3B, KCNJ8, SCN2B, ABCC9, CACNB2, KCNE3, SLMAP, KCNH2, GPD1L, HEY2, HCN4, AKAP9, CALM2, ANK2, LINC02523, KCNQ1, RYR2, VCL, MYH7, FGF11, NOS1AP, KCNE2, HARS1, GJA1, FGF14, FGF12, SEMA3A, EPHA3, BHLHE40, EGR3, DSP, DELEC1, DSG2, ATN1, XIRP1, LRRC10, HGS, NR4A3, KCNAB2, MFAP1, RRAD, ATXN2, SCD, KCNK1, APRT, SCN4B, KCNE1, KCND2, SRSF5, SKP1, TBX5, ELOC, ELOB, ELOA, IDS, SCN4A

-
Fraser Syndrome
Wikipedia
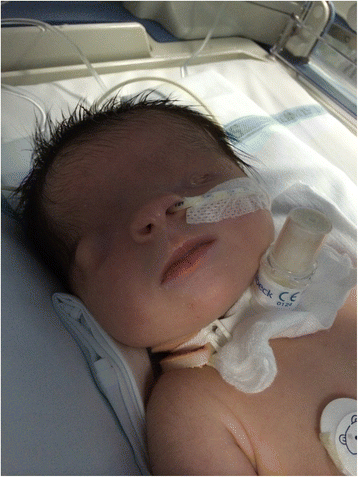
For the anatomical abnormality observed in 1964, see Frasier syndrome . Fraser syndrome Bilateral cryptophthalmos with microphthalmos in the left ocular globe and abnormal right ocular globe in a female infant with Fraser syndrome. Specialty Medical genetics Fraser syndrome (also known as Meyer-Schwickerath's syndrome , Fraser-François syndrome , or Ullrich-Feichtiger syndrome ) is an autosomal recessive congenital disorder , [1] [2] identified by several developmental anomalies. Fraser syndrome is named for the geneticist George R. ... One sib appeared to have a lethal form of ablepharon-macrostomia syndrome (AMS; 200110) or an intermediate phenotype between AMS and Fraser syndrome, and the other had classic Fraser syndrome. ... Syndrome malformatif avec cryptophthalmie.
-
Romano–ward Syndrome
Wikipedia
While other forms of long QT syndrome are associated with deafness ( Jervell and Lange-Nielsen syndrome ), intermittent weakness and bone abormailities (LQT7, Andersen-Tawil syndrome ), and autism spectrum disorder (LQT8, Timothy syndrome ), these extra-cardiac manifestations are not seen in Romano-Ward. [8] Causes [ edit ] Romano–Ward syndrome is a descriptive term for a group of subtypes of long QT syndrome, specifically subtypes LQT1-6 and LQT9-16. [8] Several subtypes of Romano–Ward syndrome have been described based on the underlying genetic variant. [5] These subtypes differ in clinical presentation and their response to treatment. ... A mutation in the ANK2 gene likely alters the flow of ions between cells in the heart, which disrupts the heart's normal rhythm and results in the features of Romano–Ward syndrome. [ medical citation needed ] Diagnosis [ edit ] Main article: Long QT syndrome [Diagnosis] The normal range of QT intervals in the normal population and in those with Romano-Ward syndrome Characteristic T-wave patterns in the 3 major subtypes of Romano-Ward syndrome Romano–Ward syndrome is principally diagnosed by measuring the QT interval corrected for heart rate (QTc) on a 12-lead electrocardiogram (ECG). Romano–Ward syndrome is associated with a prolonged QTc, although in some genetically proven cases of Romano–Ward syndrome this prolongation can be hidden, known as concealed Long QT syndrome. [12] The QTc is less than 450 ms in 95% of normal males, and less than 460 ms in 95% of normal females. Romano–Ward syndrome is suggested if the QTc is longer than these cutoffs. ... These devices are recommended for those with Romano–Ward syndrome who have experienced a cardiac arrest or a blackout whilst taking beta blockers. [8] In those who experience recurrent arrhythmias despite medical therapy, a surgical procedure called sympathetic denervation can be used to interrupt the nerves that stimulate the heart. [8] Epidemiology [ edit ] Romano–Ward syndrome is the most common form of inherited long QT syndrome, affecting an estimated 1 in 7,000 people worldwide. [ citation needed ] See also [ edit ] Long QT syndrome Jervell and Lange-Nielsen syndrome Andersen-Tawil syndrome Timothy syndrome References [ edit ] ^ a b c d e f g Tester DJ, Schwartz PJ, Ackerman MJ (2013).KCNQ1, KCNH2, CALM2, EIF2AK3, KCNQ1-AS1, DSP, KCNE1, SCN5A, KCNA5, POLR3A, KCNK3, KCNA4, RWS, KCNE2, LMNA, CD59, KCNQ4, NOS1AP, IER3IP1, SCD

-
Parinaud's Syndrome
Wikipedia
Inability to move the eyes up and down Not to be confused with Parinaud's oculoglandular syndrome . This article needs additional citations for verification . ... Unsourced material may be challenged and removed. Find sources: "Parinaud's syndrome" – news · newspapers · books · scholar · JSTOR ( February 2015 ) ( Learn how and when to remove this template message ) Parinaud's syndrome Other names Dorsal midbrain syndrome, vertical gaze palsy, upward gaze palzy, sunset sign, [1] setting-sun sign, [2] sun-setting sign, [3] sunsetting sign, [4] sunset eye sign, [5] setting-sun phenomenon [5] Specialty Neurology Parinaud's syndrome is an inability to move the eyes up and down. ... Treatment [ edit ] Treatment is primarily directed towards etiology of the dorsal midbrain syndrome. A thorough workup, including neuroimaging is essential to rule out anatomic lesions or other causes of this syndrome. ... Further reading [ edit ] Aguilar-Rebolledo F, Zárate-Moysén A, Quintana-Roldán G (1998). "Parinaud's syndrome in children". Rev. Invest. Clin. ... External links [ edit ] Classification D ICD - 10 : G46.3 ICD - 9-CM : 378.81 MeSH : D015835 DiseasesDB : 32982 v t e Symptoms , signs and syndromes associated with lesions of the brain and brainstem Brainstem Medulla (CN 8, 9, 10, 12) Lateral medullary syndrome/Wallenberg PICA Medial medullary syndrome/Dejerine ASA Pons (CN 5, 6, 7, 8) Upper dorsal pontine syndrome/Raymond-Céstan syndrome Lateral pontine syndrome ( AICA ) (lateral) Medial pontine syndrome / Millard–Gubler syndrome / Foville's syndrome ( basilar ) Locked-in syndrome Internuclear ophthalmoplegia One and a half syndrome Midbrain (CN 3, 4) Weber's syndrome ventral peduncle, PCA Benedikt syndrome ventral tegmentum, PCA Parinaud's syndrome dorsal, tumor Claude's syndrome Other Alternating hemiplegia Cerebellum Latearl Dysmetria Dysdiadochokinesia Intention tremor ) Medial Cerebellar ataxia Basal ganglia Chorea Dystonia Parkinson's disease Cortex ACA syndrome MCA syndrome PCA syndrome Frontal lobe Expressive aphasia Abulia Parietal lobe Receptive aphasia Hemispatial neglect Gerstmann syndrome Astereognosis Occipital lobe Bálint's syndrome Cortical blindness Pure alexia Temporal lobe Cortical deafness Prosopagnosia Thalamus Thalamic syndrome Other Upper motor neuron lesion Aphasia

-
Pentasomy X
Wikipedia
Pentasomy X Other names 49,XX XXX, penta X, XXXXX syndrome [1] Specialty Medical genetics Symptoms Intellectual disability , short height, low-set ears , decreased muscle tone , developmental delay [1] [2] Complications Congenital heart disease [3] Causes 5 X chromosomes [2] Risk factors Older parents [2] Diagnostic method Chromosomal analysis [2] Differential diagnosis Down syndrome , triple X syndrome , tetrasomy X , Turner syndrome [2] Treatment Based on symptoms [2] Frequency Extremely rare [2] Pentasomy X , also known as 49,XXXXX , is a chromosomal abnormality in which a female has five X chromosomes instead of the normal two. [2] Signs may include intellectual disability , short height, low-set ears , decreased muscle tone , and developmental delay . [1] [2] Complications may include congenital heart disease . [3] The condition is due to problems during the formation of the reproductive cells in a person's parents. [2] Risk factors include older parents at the time of conception . [2] Diagnosis is suspected based on symptoms and confirmed by chromosomal analysis . [2] Treatment is based on symptoms. [2] The condition is extremely rare, with fewer than forty reported cases as of 2011. [2] [3] The condition was first described in 1963. [2] Contents 1 Signs and symptoms 2 Causes 3 See also 4 References 5 External links Signs and symptoms [ edit ] The main characteristics of pentasomy X are intellectual disability , short stature and craniofacial abnormalities. [4] Other physical traits include the following: Small head [5] Ear abnormalities [5] Widely spaced eyes with upward slanting palpebral fissures and epicanthal folds [5] Short neck [5] Broad nose with a depressed nasal bridge [4] Hyperextension of the elbows [4] [5] Dental abnormalities and cleft palate [4] [5] Clinodactyly of the fifth finger [4] [5] Deformities of the feet [4] [5] Heart defects [5] [6] Causes [ edit ] The aneuploidy is thought to be caused by problems occurring during meiosis , either in the mother or in both the mother and father. Successive nondisjunctions have been observed in the mother of at least one patient. [4] [6] The features of the syndrome likely arise due to failure of X-inactivation and the presence of multiple X chromosomes from the same parent causing problems with parental imprinting . ... The reason for this is thought to be the presence of an unusually large, and imbalanced, number of X chromosomes interfering with the process. [6] See also [ edit ] Trisomy X Tetrasomy X References [ edit ] ^ a b c "49,XXXXX syndrome" . GARD . Retrieved 22 January 2018 . ^ a b c d e f g h i j k l m n o "Penta X Syndrome" . ... PMID 15333671 . ^ a b c d e f g h i Monheit, A.; Francke, U.; Saunders, B.; Jones, K. L. (1980-10-01). "The penta-X syndrome" . Journal of Medical Genetics . 17 (5): 392–396. doi : 10.1136/jmg.17.5.392 . ... External links [ edit ] Classification D ICD - 10 : Q97.1 MeSH : C535319 External resources Orphanet : 11 Tetrasomy & Pentasomy X Syndrome Information and Support v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22

-
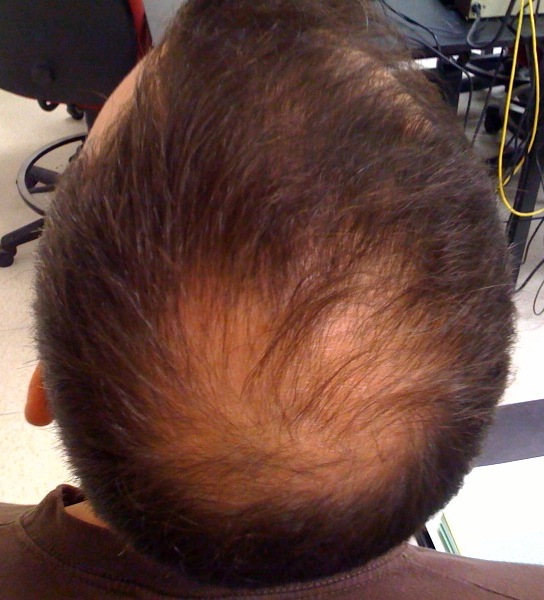
Loose Anagen Syndrome
Wikipedia
Loose anagen syndrome Other names Loose anagen hair syndrome [1] Hair loss and thinning at the back of the scalp Pronunciation / lʉːs ænədʒən ‘sɪndrəʊm / Specialty Dermatology Differential diagnosis Short anagen syndrome , alopecia areata , telogen effluvium , trichotillomania , toxic ingestion [2] Treatment Minoxidil [3] Frequency 2-2.5 cases per million people, 6:1 ratio; females to males respectively [3] [4] Loose anagen syndrome , also known as loose anagen hair syndrome, is a hair disorder related to dermatology . ... Loose anagen hair syndrome type C is most frequently seen in adults. [2] [3] There is not specific treatment for each type of loose anagen syndrome. [7] It is also unknown as to how each type is developed. [7] Differential diagnosis [ edit ] Loose Anagen Syndrome can commonly be misdiagnosed for other skin and hair disorders such as short anagen syndrome , alopecia areata , telogen effluvium , trichotillomania and toxic ingestion . ... The hairs that are removed in a telogen effluvium pull test are telogen staged hairs instead of being at the anagen stage as seen in loose anagen hair syndrome. [2] Associated conditions [ edit ] Loose anagen syndrome usually appears as an individual condition. [3] Loose anagen syndrome can be present with other conditions related to genetics or developmental conditions. [3] Examples of these conditions include; Coloboma , Noonan syndrome , Hypohidrotic ectodermal dysplasia , EEC (ectrodactyly- ectodermal dysplasia-clefting) syndrome, Neurofibromatosis , Trichorhinophalangeal syndrome , Trichotillomania , Nail-patella syndrome and Woolly hair . ... ISBN 1-4160-2999-0 . ^ a b c d e f g h i j k l m n o p q r s t u v "Loose Anagen Syndrome (pluckable hair, loose hair syndrome, loose anagen hair syndrome, syndrome of loosely attached hair in childhood)" . ... CS1 maint: multiple names: authors list ( link ) ^ Lalević-Vasić B, Polić D, Milinković R. (1990). "Le syndrome des cheveux anagènes caducs [Loose anagen hair syndrome]".
-
Yunis–varon Syndrome
Wikipedia
Yunis–Varon syndrome Other names Cleidocranial dysplasia with micrognathia, absent thumbs and distal aphalangia Yunis–Varon syndrome has an autosomal recessive pattern of inheritance . ... PMID 18203163 . S2CID 26679823 . ^ Yunis Varon Syndrome ^ a b Yunis E, Varón H (July 1980). ... PMID 20932945 . ^ a b Bhatia S, Holla RG (April 2005). "Yunis-Varon syndrome" (PDF) . Indian Pediatrics . 42 (4): 373–5. ... PMID 20932945 . ^ "Yunis Varon Syndrome" . NORD ( National Organization for Rare Disorders ) . ... External links [ edit ] Yunis-Varon syndrome; Cleidocranial dysplasia, micrognathia, absent thumbs, & distal aphalangia at NIH 's Office of Rare Diseases Classification D ICD - 10 : Q87.8 OMIM : 216340 MeSH : C536719 C536719, C536719 DiseasesDB : 33830 External resources Orphanet : 3472 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome

-
Polymalformative Genetic Syndrome With Increased Risk Of Developing Cancer
Orphanet
Polymalformative genetic syndrome with increased risk of developing cancer (PGSIRC) comprises a wide range of syndromes characterized by congenital malformations with a high risk of developing tumors including up to 50 different rare diseases. Epidemiology There are no published data on the prevalence of the set of all syndromes included in this category but, as each syndrome has its specific prevalence, it can be roughly estimated at 1/10,000. Clinical description PGSIRC encompasses different syndromes: the Overgrowth Syndromes, among which the Beckwith-Wiedemann syndrome with a 10% cumulative risk of cancer at 4 years of age mainly hepatoblastoma and Wilms' tumor, Costello syndrome with up to 10% risk of rhabdomyosarcoma, and other syndromes such as Simpson-Golabi-Behmel or Perlman (see these terms). ... The clinical characteristics are specific to each syndrome. Etiology The etiology is specific to each syndrome. ... Genetic counseling The inheritance pattern is specific to each syndrome. In most cases of familial cancer, those not associated with congenital abnormalities, tumor susceptibility is inherited in an autosomal dominant pattern.
-
Usher Syndrome
Medlineplus
Usher syndrome is a condition characterized by partial or total hearing loss and vision loss that worsens over time. ... Researchers have identified three major types of Usher syndrome, designated as types I, II, and III. ... Frequency Usher syndrome affects around 4 to 17 in 100,000 people. ... Causes Usher syndrome can be caused by mutations in several different genes. ... Usher syndrome type III is most often caused by mutations in the CLRN1 gene.
-
Short Rib–polydactyly Syndrome
Wikipedia
Short rib–polydactyly syndrome Specialty Medical genetics Short rib–polydactyly syndrome is a family of four closely related dysplasias : I – "Saldino-Noonan type" II – " Majewski type " III – "Verma-Naumoff type" (associated with DYNC2H1 ) [1] IV – "Beemer-Langer type" References [ edit ] ^ Merrill AE, Merriman B, Farrington-Rock C, et al. ... "Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome" . Am. J. Hum. Genet . 84 (4): 542–9. doi : 10.1016/j.ajhg.2009.03.015 . ... External links [ edit ] Classification D ICD - 10 : Q77.2 ICD - 9-CM : 756.5 OMIM : 263530 263520 263510 269860 MeSH : D012779 DiseasesDB : 32791 v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins This article about a disease of musculoskeletal and connective tissue is a stub .









