-
Hypertrichosis Cubiti
Wikipedia
Hypertrichosis cubiti Specialty Dermatology Hypertrichosis cubiti (also known as " hairy elbow syndrome " [1] ) is a cutaneous condition characterized by multiple terminal hairs on both elbows in children. [1] Contents 1 Causes 2 Diagnosis 3 See also 4 References Causes [ edit ] One known cause of hypertrichosis cubiti is Wiedemann-Steiner syndrome . [2] [3] Diagnosis [ edit ] This section is empty. ... "De Novo Mutations in MLL Cause Wiedemann-Steiner Syndrome" . Am J Hum Genet . 91 : 358–64. doi : 10.1016/j.ajhg.2012.06.008 . ... "De Novo Mutations in MLL Cause Wiedemann-Steiner Syndrome" . American Journal of Human Genetics . 91 (2): 358–364. doi : 10.1016/j.ajhg.2012.06.008 .
-
Chondritis
Wikipedia
See also [ edit ] Chondropathy Chondrolysis Perichondritis , commonly involving the outer ear Relapsing polychondritis References [ edit ] ^ " Chondritis " at Dorland's Medical Dictionary v t e Bone and joint disease Bone Inflammation endocrine : Osteitis fibrosa cystica Brown tumor infection : Osteomyelitis Sequestrum Involucrum Sesamoiditis Brodie abscess Periostitis Vertebral osteomyelitis Metabolic Bone density Osteoporosis Juvenile Osteopenia Osteomalacia Paget's disease of bone Hypophosphatasia Bone resorption Osteolysis Hajdu–Cheney syndrome Ainhum Gorham's disease Other Ischaemia Avascular necrosis Osteonecrosis of the jaw Complex regional pain syndrome Hypertrophic pulmonary osteoarthropathy Nonossifying fibroma Pseudarthrosis Stress fracture Fibrous dysplasia Monostotic Polyostotic Skeletal fluorosis bone cyst Aneurysmal bone cyst Hyperostosis Infantile cortical hyperostosis Osteosclerosis Melorheostosis Pycnodysostosis Joint Chondritis Relapsing polychondritis Other Tietze's syndrome Combined Osteochondritis Osteochondritis dissecans Child leg: hip Legg–Calvé–Perthes syndrome tibia Osgood–Schlatter disease Blount's disease foot Köhler disease Sever's disease spine Scheuermann's_disease arm: wrist Kienböck's disease elbow Panner disease This article about a disease of musculoskeletal and connective tissue is a stub .
-
Orofaciodigital Syndrome Vii
Omim
Although Nowaczyk et al. (2003) did not identify any mutations in the OFD1 gene (CXORF5; 300170) in these patients, they concluded that the patients had OFDS I and recommended that OFDS VII be removed from the classification of OFD syndromes. Inheritance Whelan et al. (1975) concluded that the pedigree exhibited X-linked dominant inheritance, but Munke et al. (1990) suggested that autosomal dominant inheritance was also possible.
-
Thoracopelvic Dysostosis
Omim
The disorder did not fit into any previously described syndromes such as Ellis-van Creveld syndrome (225500), short rib-polydactyly syndromes, and asphyxiating thoracic dysplasia (see 208500). ... Burn et al. (1986) suggested that the kindred reported by Bankier and Danks (1983) may have Barnes syndrome (187760). Inheritance Bankier and Danks (1983) suggested that the inheritance pattern of thoracopelvic dysostosis is autosomal dominant.
-
Larynx, Congenital Partial Atresia Of
Omim
They studied a family in which 3 members had the cri du chat syndrome (123450) with deletion only of the distal half of the light band 5p15. As patients with the cri-du-chat syndrome age, about 30% of them develop gray hair prematurely (Niebuhr, 1971). ... Overhauser et al. (1994) concluded that a critical chromosomal region involved in the high-pitched cry of the cat cry syndrome is 5p15.3, while the chromosomal region involved in the remaining features of the syndrome is a small region within 5p15.2.
-
Mental Retardation, X-Linked, Syndromic 12
Omim
Mapping By linkage analysis using probes for DXS255 located at Xp11.22 in a family segregating a syndromic form of mental retardation, Wilson et al. (1992) obtained a maximum 2-point lod score of 2.10 if phase was inferred and 1.20 if it was not.
-
Acute Respiratory Distress Syndrome
Gard
Acute respiratory distress syndrome (ARDS) is a life-threatening lung condition that prevents enough oxygen from getting to the lungs and into the blood.THBD, EDN1, RAMP2, SOD2, CCL2, ALB, MT3, CHPT1, MIR30B, ACAA2, MIR30A, MIR99A, MIR135B, MIR337, IGFBP6, IFRD1, MIR346, GNRH1, MIR380, TIMP1, GABRB1, MIRLET7B, MIR215, FGFR4, CYB5A, MIR210, MIR19A, MIR18A, TSPAN8, MIR128-2, MIR127, MIR126, PRDX6, MIRLET7D, FBLN5, EBF1, SERPINH1, LIM2, SLC25A11, CES3, ACO2, PLA2G4A, PROS1, PSMA4, EIF2AK1, ALAD, AKR1B1, MT2A, APC, S100A4, S100A9, MDH1, LCT, LCN2, SERPINC1, IL10, AGER, IL1RN, CXCL10, HSPA4, PDPN, TBXA2R, C3AR1, MIR26A1, MFN1, C5AR1, C5, CXCL8, SFTPB, TNF, BTBD8, IL6, ABCA3, TGFB1, ACE, VEGFA, ANGPT2, SFTPD, IL17A, ACE2, SFTPC, SFTPA2, IL1B, MMP9, NFE2L2, SFTPA1, HMOX1, HMGB1, NAMPT, TLR4, IL18, MPO, NLRP3, AGT, IL1A, FGF7, REN, NR3C1, VPS51, GABPA, SCGB1A1, AGTR1, PI3, MIR155, ELANE, STAT3, SARS2, IFNG, ANGPT1, IGF1, LINC01672, MIF, PLA2G7, SARS1, MAPK1, CRP, HP, YWHAZ, PLA2G1B, SMUG1, F3, PLB1, CFTR, POLDIP2, MAPK14, IL33, FAS, VWF, NFKB1, FASLG, CXCL5, MBL2, MYLK, AIMP2, IL17D, SIRT3, SIRT1, PLA2G2A, PIK3CB, SERPINA1, SERPINE1, RNF19A, PTGS2, NT5E, NOS3, HPGDS, ABCA4, AHSA1, MOK, RELA, LTA, SLC12A2, HABP2, STAT5A, IL27, TLR3, CBLIF, TM7SF2, UCN3, GZMB, DCN, MYDGF, TFPI, HGF, TEK, CRK, SYT1, MIR223, STAT5B, ICAM1, GORASP1, ARSD, ELAVL1, TSPO, EPAS1, PROCR, MIR34A, PLF, CHRM3, WNK1, GRAP2, CCN2, HAP1, KCNQ1OT1, PIEZO1, LATS1, ATG5, SLC27A5, IFI44L, PCYT1B, MAPKAPK2, BMS1, AKT3, HCST, ITM2B, ARID5A, CCNB2, HPSE, EBI3, CELF2, KLF2, ADIPOQ, ISG15, SH2B2, KL, RIPK3, AKR1B10, CNMD, RAMAC, LINC01194, NPSR1, H3P44, TREML1, OR10A4, HCA1, ANKS4B, XKR3, SERPINA12, CCDC26, INO80C, IL17RC, MINDY4, KAT8, TAS1R3, MIR122, MIR150, MIR211, MIR615, RN7SL263P, RNU6-392P, LARP1BP2, THRIL, AD14, MIR802, MIR506, MIR216A, MIR494, MIR146B, MIR381, MIR27B, MIR27A, MIR221, PARP9, SLC2A10, POLG2, PANK2, ANLN, TREM1, TLR8, CLEC1B, ANGPTL4, NOX4, SMARCAL1, NAAA, NOX1, HAVCR1, SPATS2L, IL17RA, LY96, KPNA6, ANGPTL2, EGLN1, UGT1A1, PPP6R3, POLD4, DOK3, LPCAT1, GSDMD, CORO7, POPDC3, ZNF335, PELI2, CARMIL1, HDAC3, POLE4, IL36G, NSMCE3, SLC50A1, CENPJ, TIMELESS, RAB27A, PIK3R3, ETS2, FOS, FN1, FOXM1, FER, FAAH, F5, F2, ERG, IL2, EPHX2, EPHB2, EGF, S1PR3, DPP4, DNASE1, NQO1, FUSE, GAD1, GJA5, CXCR3, IKBKB, IGFBP3, IFNB1, IFIT1, TNC, HSPA1B, HSPA1A, HSD11B2, HRG, HNRNPA1, FOXA2, HIF1A, HADHA, GZMA, GPT, CFD, DEFB1, DAPK1, CD40, CCNB1, CAV1, SERPING1, BTK, BMP7, AQP5, AQP1, KLK3, BIRC3, APCS, ANXA2, AGTR2, ADCY9, ACACA, ABO, CD14, CD40LG, CYP3A5, CD44, CYP3A7, CYP1B1, CYP1A1, CTSG, CTNNB1, CST3, CRYGD, COL4A5, CCR4, CIRBP, CEL, CDKN3, CDH5, CD68, CD59, IL1R1, IL2RA, PLA2G10, PTK2, SMPD1, SLC22A4, SELPLG, CCL7, RENBP, PRPH2, PTN, PSMD12, IL4, PROS2P, PROC, MAP2K1, MAPK8, PPARA, POMC, FXYD1, FSCN1, ST2, TAPBP, TCF21, FGF23, ARHGEF5, CXCR4, XDH, VIP, UGT8, TXNRD1, TXN, TST, TPI1, TOP2A, TNFRSF1A, NKX2-1, TIA1, TERT, PLD1, PLCL1, PLAUR, MMP3, CXCL9, MAP3K1, MDK, MCL1, SMAD3, SH2D1A, LY6E, LHCGR, LGALS3, KRT19, KDR, ITGB2, ITGAM, IL13, IL7R, MMP2, MPP1, PIK3CG, MUC1, PIK3CD, PIK3CA, CFP, PRDX1, PAFAH1B1, P4HB, P2RX7, OPRM1, OAS3, NM, NFYA, NFKBIA, MYD88, MYC, MUC5AC, H3P5

-
Peroxisome Biogenesis Disorder-Zellweger Syndrome Spectrum
Gard
The spectrum includes Zellweger syndrome (ZS), the most severe form; neonatal adrenoleukodystrophy (NALD), an intermediate form; and infantile Refsum disease (IRD), the least severe form.
-
Al-Gazali Syndrome
Omim
Of the 4 live births, only 2 were affected with clinical features of al-Gazali syndrome. The affected boys had intrauterine growth retardation, bilateral talipes equinovarus, contractures of the large joints, camptodactyly, multiple fractures, and abnormalities of the anterior segment of the eyes.
-
Reactive Airway Disease
Wikipedia
Unlike RAD, reactive airways dysfunction syndrome is recognized by multiple societies as a real clinical syndrome, including the American Thoracic Society and the American College of Chest Physicians . [1] Contents 1 Terminology 2 Reactive airways dysfunction syndrome 3 Controversy over use 4 See also 5 References 6 External links Terminology [ edit ] It is common to see RAD incorrectly used as a synonym for asthma The term reactive airway disease originally began to appear in medical literature in the 1980s in reference to asthmatic patients with hyperactive airways, which is a common feature of asthma. ... If none of these symptoms are present in an adult patients' medical history or documentation, the physician may label the patient with RAD instead of asthma in order to still indicate there is an airway issue without formal diagnosis. [1] Reactive airways dysfunction syndrome [ edit ] While the acronyms are similar, reactive airway disease (RAD) and reactive airways dysfunction syndrome (RADS) are not the same. [1] Reactive airways dysfunction syndrome was first identified by Stuart M. ... While some physicians argue that RADS is also not a real clinical syndrome, it is more commonly recognized in legitimate associations than RAD. ... Bernstein (1985). "Reactive airways dysfunction syndrome (RADS): persistent asthma syndrome after high level irritant exposures". ... "Which agents cause reactive airways dysfunction syndrome (RADS)? A systematic review" .
-
Endocardial Fibroelastosis
Wikipedia
It is an uncommon cause of unexplained heart failure in infants and children, and is one component of HEC syndrome . Fibroelastosis is strongly seen as a primary cause of restrictive cardiomyopathy in children, along with cardiac amyloidosis, which is more commonly seen in progressive multiple myeloma patients and the elderly. [ citation needed ] Cause [ edit ] A review cites references to 31 different diseases and other stresses associated with the EFE reaction. [2] These include infections, cardiomyopathies, immunologic diseases, congenital malformations, even electrocution by lightning strike. ... Now there are specific named genes associated with certain cardiomyopathies, some of which show the characteristic reaction of EFE. A typical example is Barth syndrome and the responsible gene, tafazzin. [13] Developments in echocardiography, both the technology of the machines and the skill of the operators, have made it no longer necessary to see the endocardium at autopsy. ... PMID 8994428 . ^ Bione S, D'Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D (1996) "A novel x-linked gene, G4.5, is responsible for Barth Syndrome" Nat Genet 12: 385-389. ^ Rustico MA, Benettoni A, Bussani R, Maieron A, Mandruzzato G. (1995) "Early fetal endocardial fibroelastosis and critical aortic stenosis: a case report" Ultrasound Obstet Gynecol 5: 202-205. ^ Raboisson M-J, Fouron J-C, Sonesson S-E, Nyman M, Proulx F, Gamache S (2005) " Fetal Doppler echocardiographic diagnosis and successful steroid therapy of Luciani-Wenckebach phenomenon and endocardial fibroelastosis related to maternal anti-Ro and anti-La antibodies" J Am Soc Echocardiogr 18: 375-380. ^ Alvarez JA, Wilkinson JD, Lipshultz SE (2007) "Outcome predictors for pediatric dilated cardiomyopathy: a systematic review" Prog Pediatr Cardiol 23: 25-32. External links [ edit ] Classification D ICD - 10 : I42.4 ICD - 9-CM : 425.3 OMIM : 226000 305300 MeSH : D004695 DiseasesDB : 29236 External resources Orphanet : 2022 v t e Cardiovascular disease (heart) Ischaemic Coronary disease Coronary artery disease (CAD) Coronary artery aneurysm Spontaneous coronary artery dissection (SCAD) Coronary thrombosis Coronary vasospasm Myocardial bridge Active ischemia Angina pectoris Prinzmetal's angina Stable angina Acute coronary syndrome Myocardial infarction Unstable angina Sequelae hours Hibernating myocardium Myocardial stunning days Myocardial rupture weeks Aneurysm of heart / Ventricular aneurysm Dressler syndrome Layers Pericardium Pericarditis Acute Chronic / Constrictive Pericardial effusion Cardiac tamponade Hemopericardium Myocardium Myocarditis Chagas disease Cardiomyopathy Dilated Alcoholic Hypertrophic Tachycardia-induced Restrictive Loeffler endocarditis Cardiac amyloidosis Endocardial fibroelastosis Arrhythmogenic right ventricular dysplasia Endocardium / valves Endocarditis infective endocarditis Subacute bacterial endocarditis non-infective endocarditis Libman–Sacks endocarditis Nonbacterial thrombotic endocarditis Valves mitral regurgitation prolapse stenosis aortic stenosis insufficiency tricuspid stenosis insufficiency pulmonary stenosis insufficiency Conduction / arrhythmia Bradycardia Sinus bradycardia Sick sinus syndrome Heart block : Sinoatrial AV 1° 2° 3° Intraventricular Bundle branch block Right Left Left anterior fascicle Left posterior fascicle Bifascicular Trifascicular Adams–Stokes syndrome Tachycardia ( paroxysmal and sinus ) Supraventricular Atrial Multifocal Junctional AV nodal reentrant Junctional ectopic Ventricular Accelerated idioventricular rhythm Catecholaminergic polymorphic Torsades de pointes Premature contraction Atrial Junctional Ventricular Pre-excitation syndrome Lown–Ganong–Levine Wolff–Parkinson–White Flutter / fibrillation Atrial flutter Ventricular flutter Atrial fibrillation Familial Ventricular fibrillation Pacemaker Ectopic pacemaker / Ectopic beat Multifocal atrial tachycardia Pacemaker syndrome Parasystole Wandering atrial pacemaker Long QT syndrome Andersen–Tawil Jervell and Lange-Nielsen Romano–Ward Cardiac arrest Sudden cardiac death Asystole Pulseless electrical activity Sinoatrial arrest Other / ungrouped hexaxial reference system Right axis deviation Left axis deviation QT Short QT syndrome T T wave alternans ST Osborn wave ST elevation ST depression Strain pattern Cardiomegaly Ventricular hypertrophy Left Right / Cor pulmonale Atrial enlargement Left Right Athletic heart syndrome Other Cardiac fibrosis Heart failure Diastolic heart failure Cardiac asthma Rheumatic fever v t e Congenital heart defects Heart septal defect Aortopulmonary septal defect Double outlet right ventricle Taussig–Bing syndrome Transposition of the great vessels dextro levo Persistent truncus arteriosus Aortopulmonary window Atrial septal defect Sinus venosus atrial septal defect Lutembacher's syndrome Ventricular septal defect Tetralogy of Fallot Atrioventricular septal defect Ostium primum Consequences Cardiac shunt Cyanotic heart disease Eisenmenger syndrome Valvular heart disease Right pulmonary valves stenosis insufficiency absence tricuspid valves stenosis atresia Ebstein's anomaly Left aortic valves stenosis insufficiency bicuspid mitral valves stenosis regurgitation Other Underdeveloped heart chambers right left Uhl anomaly Dextrocardia Levocardia Cor triatriatum Crisscross heart Brugada syndrome Coronary artery anomaly Anomalous aortic origin of a coronary artery Ventricular inversion
-
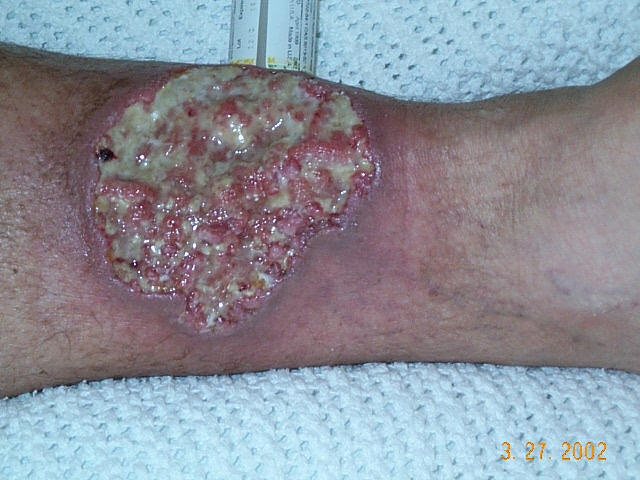
Pyoderma Gangrenosum
Wikipedia
Though it can affect people of any age, it mostly affects people in their 40s and 50s. [1] Contents 1 Types 2 Presentation 2.1 Associations 3 Causes 4 Diagnosis 4.1 Diagnostic criteria 5 Treatment 6 See also 7 References 8 External links Types [ edit ] Pyoderma gangrenosum There are two main types of pyoderma gangrenosum: [1] the 'typical' ulcerative form, which occurs in the legs an 'atypical' form that is more superficial and occurs in the hands and other parts of the body Other variations are: [4] Peristomal pyoderma gangrenosum comprises 15% of all cases of pyoderma Bullous pyoderma gangrenosum Pustular pyoderma gangrenosum [5] Vegetative pyoderma gangrenosum [6] Presentation [ edit ] Associations [ edit ] The following are conditions commonly associated with pyoderma gangrenosum: [7] Inflammatory bowel disease: Ulcerative colitis Crohn's disease Arthritides: Rheumatoid arthritis Seronegative arthritis Hematological disease: Myelocytic leukemia [8] Hairy cell leukemia Myelofibrosis Myeloid metaplasia Monoclonal gammopathy Autoinflammatory disease: Pyogenic sterile arthritis, and acne syndrome ( PAPA syndrome ) Causes [ edit ] Though the cause is not well understood, the disease is thought to be due to immune system dysfunction, and particularly improper functioning of neutrophils . In support of an immune cause, a variety of immune mediators such as interleukin (IL)-8 , IL-1β , IL-6 , interferon (IFN)-γ , granulocyte colony-stimulating factor , tumor necrosis factor alpha , matrix metalloproteinase (MMP)-9 , MMP10 , and elafin have all been reported to be elevated in patients with pyoderma gangrenosum. [9] Also in support of an immune cause is the finding that at least half of all pyoderma gangrenosum patients suffer from immune-mediated diseases. [1] For instance, ulcerative colitis , rheumatoid arthritis , and multiple myeloma (MM) have all been associated with pyoderma gangrenosum. It can also be part of a syndromes such as PAPA syndrome . [ citation needed ] One hallmark of pyoderma gangrenosum is pathergy , which is the appearance of new lesions at sites of trauma, including surgical wounds. [10] Diagnosis [ edit ] Diagnosis of PG is challenging owing to its variable presentation, clinical overlap with other conditions, association with several systemic diseases, and absence of defining histopathologic or laboratory findings. ... External links [ edit ] Classification D ICD - 10 : L88 ICD - 9-CM : 686.01 MeSH : D017511 DiseasesDB : 11064 SNOMED CT : 74578003 External resources eMedicine : article/1123821 Orphanet : 48104 Wikimedia Commons has media related to Pyoderma gangrenosum . v t e Neutrophilic and eosinophilic dermatoses Eosinophilic dermatosis With vasculitis Eosinophilic vasculitis Eosinophilic granulomatosis with polyangiitis Without vasculitis Arthropod assault Eosinophilic cellulitis Hypereosinophilic syndrome Papuloerythroderma of Ofuji Granuloma faciale Eosinophilic folliculitis Ungrouped Angiolymphoid hyperplasia with eosinophilia / Kimura's disease Annular erythema of infancy Eosinophilic fasciitis Eosinophilic granuloma Eosinophilic ulcer of the oral mucosa Erythema toxicum neonatorum Incontinentia pigmenti Itchy red bump disease Juvenile xanthogranuloma Pachydermatous eosinophilic dermatitis Papular eruption of blacks Pruritic papular eruption of HIV disease Reactive neutrophilic dermatoses Epidermis Keratoderma blennorrhagicum Subcorneal pustular dermatosis Dermis without vasculitis : Sweet's syndrome Pyoderma gangrenosum Bowel-associated dermatosis–arthritis syndrome with vasculitis: Neutrophilic dermatosis of the dorsal hands Ungrouped Acute erythema nodosum Marshall syndrome Neutrophilic eccrine hidradenitis Pyogenic arthritis–pyoderma gangrenosum–acne syndrome Rheumatoid neutrophilic dermatitis Superficial granulomatous pyoderma Sweet's syndrome-like dermatosis Vesicopustular dermatosis v t e Cutaneous keratosis, ulcer, atrophy, and necrobiosis Epidermal thickening keratoderma : Keratoderma climactericum Paraneoplastic keratoderma Acrokeratosis paraneoplastica of Bazex Aquagenic keratoderma Drug-induced keratoderma psoriasis Keratoderma blennorrhagicum keratosis : Seborrheic keratosis Clonal seborrheic keratosis Common seborrheic keratosis Irritated seborrheic keratosis Seborrheic keratosis with squamous atypia Reticulated seborrheic keratosis Dermatosis papulosa nigra Keratosis punctata of the palmar creases other hyperkeratosis : Acanthosis nigricans Confluent and reticulated papillomatosis Callus Ichthyosis acquisita Arsenical keratosis Chronic scar keratosis Hyperkeratosis lenticularis perstans Hydrocarbon keratosis Hyperkeratosis of the nipple and areola Inverted follicular keratosis Lichenoid keratosis Multiple minute digitate hyperkeratosis PUVA keratosis Reactional keratosis Stucco keratosis Thermal keratosis Viral keratosis Warty dyskeratoma Waxy keratosis of childhood other hypertrophy: Keloid Hypertrophic scar Cutis verticis gyrata Necrobiosis / granuloma Necrobiotic/palisading Granuloma annulare Perforating Generalized Subcutaneous Granuloma annulare in HIV disease Localized granuloma annulare Patch-type granuloma annulare Necrobiosis lipoidica Annular elastolytic giant-cell granuloma Granuloma multiforme Necrobiotic xanthogranuloma Palisaded neutrophilic and granulomatous dermatitis Rheumatoid nodulosis Interstitial granulomatous dermatitis / Interstitial granulomatous drug reaction Foreign body granuloma Beryllium granuloma Mercury granuloma Silica granuloma Silicone granuloma Zirconium granuloma Soot tattoo Tattoo Carbon stain Other/ungrouped eosinophilic dermatosis Granuloma faciale Dermis / localized CTD Cutaneous lupus erythematosus chronic: Discoid Panniculitis subacute : Neonatal ungrouped: Chilblain Lupus erythematosus–lichen planus overlap syndrome Tumid Verrucous Rowell's syndrome Scleroderma / Morphea Localized scleroderma Localized morphea Morphea–lichen sclerosus et atrophicus overlap Generalized morphea Atrophoderma of Pasini and Pierini Pansclerotic morphea Morphea profunda Linear scleroderma Atrophic / atrophoderma Lichen sclerosus Anetoderma Schweninger–Buzzi anetoderma Jadassohn–Pellizzari anetoderma Atrophoderma of Pasini and Pierini Acrodermatitis chronica atrophicans Semicircular lipoatrophy Follicular atrophoderma Linear atrophoderma of Moulin Perforating Kyrle disease Reactive perforating collagenosis Elastosis perforans serpiginosa Perforating folliculitis Acquired perforating dermatosis Skin ulcer Pyoderma gangrenosum Other Calcinosis cutis Sclerodactyly Poikiloderma vasculare atrophicans Ainhum / Pseudo-ainhum v t e Paraneoplastic syndromes Endocrine Hypercalcaemia SIADH Zollinger–Ellison syndrome Cushing's syndrome Hematological Multicentric reticulohistiocytosis Nonbacterial thrombotic endocarditis Neurological Paraneoplastic cerebellar degeneration Encephalomyelitis Limbic encephalitis Opsoclonus Polymyositis Transverse myelitis Lambert–Eaton myasthenic syndrome Anti-NMDA receptor encephalitis Musculoskeletal Dermatomyositis Hypertrophic osteopathy Mucocutaneous reactive erythema Erythema gyratum repens Necrolytic migratory erythema papulosquamous Acanthosis nigricans Ichthyosis acquisita Acrokeratosis paraneoplastica of Bazex Extramammary Paget's disease Florid cutaneous papillomatosis Leser-Trélat sign Pityriasis rotunda Tripe palms Other Febrile neutrophilic dermatosis Pyoderma gangrenosum Paraneoplastic pemphigusPSTPIP1, MEFV, NOD2, NLRP3, TCIRG1, SYN3, ELANE, GFI1, SRP54, GLI3, TNF, PAPPA, MTHFR, MAP6, JAK2, RAG1, MPO, JAK1, INSRR, DERL1, IL25, CD40, NELFCD, IL37, ACAD8, TRAF3IP2, CD40LG, USP15, CCR3, COL17A1, TYK2, GYPA, TIMP3, TG, IL5, SOAT1, CXCL8, PTPN6, CXCR1, NM, CXCR2, PRDM1, IL15, IL17A, LAD1, JAK3, ALB
-
Minimal Change Disease
Wikipedia
Specialty Nephrology Minimal change disease (also known as MCD , minimal change glomerulopathy , and nil disease , among others ) is a disease affecting the kidneys which causes a nephrotic syndrome . [1] Nephrotic syndrome leads to the loss of significant amounts of protein in the urine, which causes the widespread edema (soft tissue swelling) and impaired kidney function commonly experienced by those affected by the disease. [1] It is most common in children and has a peak incidence at 2 to 6 years of age. [2] MCD is responsible for 10-25% of nephrotic syndrome cases in adults. [3] It is also the most common cause of nephrotic syndrome of unclear cause (idiopathic) in children. [3] Contents 1 Signs and symptoms 2 Pathology 3 Pathophysiology 3.1 Proteinuria 3.2 Edema 4 Diagnosis 5 Treatment 5.1 Children 5.2 Adults 6 Prognosis 6.1 Children 6.2 Adults 7 Epidemiology 8 Etymology 9 References 10 External links Signs and symptoms [ edit ] The clinical signs of minimal change disease are proteinuria (abnormal excretion of proteins, mainly albumin , into the urine), edema (swelling of soft tissues as a consequence of water retention), weight gain, and hypoalbuminaemia (low serum albumin). [1] These signs are referred to collectively as nephrotic syndrome . [1] Periorbital edema that can be seen in minimal change disease The first clinical sign of minimal change disease is usually edema with an associated increase in weight. [1] The swelling may be mild but patients can present with edema in the lower half of the body, periorbital edema , swelling in the scrotal/labial area and anasarca in more severe cases. [1] In older adults, patients may also present with acute kidney injury (20-25% of affected adults) and high blood pressure . [4] Due to the disease process, patients with minimal change disease are also at risk of blood clots and infections. [4] Pathology [ edit ] For years, pathologists found no changes when viewing kidney biopsy specimens under light microscopy , hence the name "minimal change disease." ... As a result of the excess fluid, individuals with minimal change disease often gain weight, as they are excreting less water in the urine, and experience fatigue. [1] Diagnosis [ edit ] As minimal change disease is a subset of nephrotic syndrome, diagnosis involves looking for a combination of edema, high amounts of protein in urine, low albumin and high serum cholesterol. Initial workup can include a urinalysis , kidney function tests , serum albumin level and a lipid panel . [6] Microscopic amounts of blood are present in the urine of 10-30% adults with MCD. [3] As MCD is the most common type of nephrotic syndrome in children, renal biopsy is not usually done in children under the age of 10 unless there are concerning features that are unusual for the disease ( high blood pressure , bloody urine , renal dysfunction ) and if they fail to respond to corticosteroid therapy. [1] These would suggest that it may not be minimal change disease. ... It is by far the most common cause of nephrotic syndrome in children, accounting for 70-90% of children >1 year of age. [4] After puberty, it is caused by minimal change disease about half the time. [4] Among young children, boys seem to be more likely to develop minimal change disease than girls (about 2:1). [1] Minimal change disease is seen in about 16 in every 100,000 children, being more common in South Asians and Native Americans, but rarer in African Americans. [1] In adults, it accounts for less than 15% of adults diagnosed with nephrotic syndrome. [4] Etymology [ edit ] Minimal change disease has been called by many other names in the medical literature, including minimal change nephropathy, minimal change nephrosis, minimal change nephrotic syndrome, minimal change glomerulopathy, foot process disease (referring to the foot processes of the podocytes ), nil disease (referring to the lack of pathologic findings on light microscopy), nil lesions, lipid nephrosis, and lipoid nephrosis. ... "Management of childhood onset nephrotic syndrome". Pediatrics . 124 (2): 747–57. doi : 10.1542/peds.2008-1559 .

-
Spontaneous Bacterial Peritonitis
Wikipedia
Where there is a risk of kidney malfunction developing in a syndrome called hepatorenal syndrome intravenous albumin is usually administered too. ... "Predictors of peritonitis in children with nephrotic syndrome". Pediatr. Nephrol . 17 (8): 678–82. doi : 10.1007/s00467-002-0890-6 . ... "Spontaneous bacterial peritonitis as a presenting feature of nephrotic syndrome". Journal of Paediatrics and Child Health (Review). 49 (12): 1069–71. doi : 10.1111/jpc.12389 . ... "Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis". ... A relatively common but rarely recognized syndrome". Ann. Intern. Med . 60 (4): 568–80. doi : 10.7326/0003-4819-60-4-568 .
-
Gastrointestinal Disease
Wikipedia
Peutz–Jeghers syndrome can cause dark spots on the oral mucosa or on the lips or the skin around the mouth. ... Peptic ulcers are also common in the duodenum. [3] : 879–884 Chronic diseases of malabsorption may affect the small intestine, including the autoimmune coeliac disease , infective Tropical sprue , and congenital or surgical short bowel syndrome . Other rarer diseases affecting the small intestine include Curling's ulcer , blind loop syndrome , Milroy disease and Whipple's disease . ... Functional colonic diseases refer to disorders without a known cause, including irritable bowel syndrome and intestinal pseudoobstruction . ... In severe cases, pancreatitis may lead to rapid blood loss and systemic inflammatory response syndrome . When the pancreas is unable to secrete digestive enzymes , such as with a pancreatic cancer occluding the pancreatic duct , result in jaundice. ... Cite journal requires |journal= ( help ) External links [ edit ] Classification D ICD - 10 : K00 - K93 MeSH : D004066 External resources MedlinePlus : 007447 v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum PneumoperitoneumALDH1A2, BARX1, LRRC8D, CRTC1, CFTR, NR1H4, HOTAIR, HULC, MIR369, OLFM4, CASR, STK11, CCL7, PPARG, MUTYH, SMAD7, DEFB1, DBN1, CCAT1

-
De Quervain Syndrome
Wikipedia
De Quervain syndrome involves noninflammatory thickening of the tendons and the synovial sheaths that the tendons run through. ... The modified Eichoff maneuver , commonly called the Finkelstein's test , is a physical exam maneuver used to diagnose de Quervain syndrome. [2] To perform the test, the examiner grasps and ulnar deviates the hand when the person has their thumb held within their fist. [15] [2] If sharp pain occurs along the distal radius (top of forearm, about an inch below the wrist), de Quervain's syndrome is likely. While a positive Finkelstein's test is often considered pathognomonic for de Quervain syndrome, the maneuver can also cause pain in those with osteoarthritis at the base of the thumb. [2] Differential diagnosis [ edit ] Differential diagnoses [16] include: Osteoarthritis of the first carpo-metacarpal joint Intersection syndrome —pain will be more towards the middle of the back of the forearm and about 2–3 inches below the wrist Wartenberg's syndrome Treatment [ edit ] As with many musculoskeletal conditions, the management of de Quervain's disease is determined more by convention than scientific data. ... The medical name for the condition is De Quervain syndrome and is associated with the tendons connected to the thumb through the wrist. ... "Hand pain other than carpal tunnel syndrome (CTS): the role of occupational factors".

-
Spastic Paraparesis-Deafness Syndrome
Orphanet
A rare neurologic disease characterized by spastic paraparesis presenting in late childhood and hearing loss. Additional features may include retinal anomalies, lenticular opacities, short stature, hypogonadism, sensory deficits, tremor, dysdiochokinesia, elevated cerebrospinal fluid protein, and absent or prolonged somatosensory evoked potentials. Plasma and fibroblast levels of saturated very long-chain fatty acids are normal. There have been no further descriptions in the literature since 1986.
-
5p13 Microduplication Syndrome
Orphanet
A rare partial autosomal trisomy/tetrasomy characterized by global developmental delay, intellectual disability, autistic behavior, muscular hypotonia, macrocephaly and facial dysmorphism (frontal bossing, short palpebral fissures, low set, dysplastic ears, short or shallow philtrum, high arched or narrow palate, micrognathia). Other associated clinical features include sleep disturbances, seizures, aplasia/hypoplasia of the corpus callosum, skeletal abnormalities (large hands and feet, long fingers and toes, talipes).
-
Multifocal Lymphangioendotheliomatosis-Thrombocytopenia Syndrome
Orphanet
A rare lymphatic system anomaly characterized by multifocal congenital and progressive vascular lesions of the skin, gastrointestinal tract, and occasionally other anatomic sites, causing potentially life-threatening thrombocytopenic coagulopathy. Macroscopically, the lesions appear as round to oval, red-brown plaques, as large as a few centimeters in diameter. Histopathologically, they consist of dilated, thin-walled vessels with variable endothelial hyperplasia, positive for lymphatic endothelial cell markers, and resembling benign lymphangioendothelioma.
-
Progeria-Short Stature-Pigmented Nevi Syndrome
Orphanet
Progeria-short stature-pigmented nevi is a progeroid disorder characterised by low birthweight, short stature, multiple pigmented nevi and lack of facial subcutaneous fat. Epidemiology Less than ten cases have been described so far. Clinical description Other clinical features include immunodeficiency, high-pitched voice and progressive hearing and visual loss. Motor and intellectual development is normal or slightly impaired.




