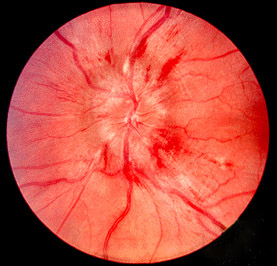
Papilledema is a condition in which increased pressure in or around the brain (intracranial pressure) causes swelling of the part of the optic nerve inside the eye ( optic disc ). Symptoms of increased intracranial pressure include headache or nausea and vomiting. Vision problems are not common initially, but may include short flickers of gray vision, blurred or double vision, and decreased field of vision or ability to see colors. Both eyes are usually affected. Papilledema by definition is caused by increased cranial pressure. Diagnosis includes a thorough eye exam by an ophthalmologist. Brain imaging studies (for example CT scan or MRI) are used to find the cause of the increased intracranial pressure.
Chromosomal disorder in which there are three copies of every chromosome Triploid syndrome Other names 69,XXX [1] 2n/3n mixoploidy, 3n syndrome, chromosome triploidy syndrome, diploid/triploid mixoploidy, triploidy, triploidy syndrome A karyotype of a person with triploidy Specialty Medical genetics Triploid syndrome , also called triploidy , is a chromosomal disorder in which a fetus has three copies of every chromosome instead of the normal two.
Triploidy is a chromosome abnormality that occurs when there is an extra set of chromosomes present in each cell. Most pregnancies affected by triploidy are lost through early miscarriage . However, reports exist of some affected babies living up to five months. Those that survive are often mosaic . The signs and symptoms associated with triploidy vary but may include a variety of birth defects and an unusually small size. This condition does not run in families and is not associated with maternal or paternal age.
Triploidy is a rare chromosomal anomaly, polyploidy, characterized by early in utero growth restriction, and multiple birth defects, including neural tube defects, facial abnormalities, cleft lip/palate, congenital heart anomalies, genital malformations, and peripheral skeletal abnormalities. It is usually prenatally lethal.
In a minority of patients, MB is associated with Gorlin syndrome, familial adenomatous polyposis (FAP; the association of FAP and MB is referred to as the Turcot syndrome with polyposis) or with Li-Fraumeni Syndrome. Increased susceptibility to certain tumors (neuroblastoma), hematological malignancies (acute lymphoblastic leukemia, acute myeloid leukemia) or disorders caused by mutations in genes encoding components of the RAS signaling pathway (Noonan syndrome or neurofibromatosis-Noonan syndrome) have been reported in MB. ... Genetic counseling Genetic counseling is indicated in specific constellations, e.g. in sonic hedgehoc-activated MB (Gorlin-syndrome, Li-Fraumeni-Syndrome, BRCA2), CTNNB1-negative WNT-activated MB (Turcot-syndrome).
In 1 to 2% of patients, medulloblastoma is associated with Gorlin syndrome (109400), a nevoid basal carcinoma syndrome. Medulloblastoma also occurs in up to 40% of patients with Turcot syndrome (276300). Medulloblastoma is thought to arise from neural stem cell precursors in the granular cell layer of the cerebellum. ... Cerebellar medulloblastoma is a feature of basal cell nevus syndrome (109400), von Hippel-Lindau syndrome (193300), and familial adenomatous polyposis (175100). ... No obvious physical stigmata of nevoid basal cell carcinoma syndrome was found among 21 mutation carriers from both families who were examined, including 11 patients who underwent brain MRI.
Classic medulloblastoma is a histological variant of medulloblastoma (see this term) ,an embryonic malignancy, having a midline location, occurring most often in children and manifesting with variable symptoms such as headaches, nausea, vomiting and ataxia.
The disorder is no longer listed in the 11th revision of the International Statistical Classification of Diseases and Related Health Problems, or ICD-11 Disorganized schizophrenia was thought to be an extreme expression of the disorganization syndrome that has been hypothesized to be one aspect of a three-factor model of symptoms in schizophrenia, [1] the other factors being reality distortion (involving delusions and hallucinations) and psychomotor poverty (lack of speech, lack of spontaneous movement and various aspects of blunting of emotion). ... External links [ edit ] Classification D ICD - 10 : F20.1 ICD - 9-CM : 295.1 MeSH : D012562 v t e Mental and behavioral disorders Adult personality and behavior Gender dysphoria Ego-dystonic sexual orientation Paraphilia Fetishism Voyeurism Sexual maturation disorder Sexual relationship disorder Other Factitious disorder Munchausen syndrome Intermittent explosive disorder Dermatillomania Kleptomania Pyromania Trichotillomania Personality disorder Childhood and learning Emotional and behavioral ADHD Conduct disorder ODD Emotional and behavioral disorders Separation anxiety disorder Movement disorders Stereotypic Social functioning DAD RAD Selective mutism Speech Stuttering Cluttering Tic disorder Tourette syndrome Intellectual disability X-linked intellectual disability Lujan–Fryns syndrome Psychological development ( developmental disabilities ) Pervasive Specific Mood (affective) Bipolar Bipolar I Bipolar II Bipolar NOS Cyclothymia Depression Atypical depression Dysthymia Major depressive disorder Melancholic depression Seasonal affective disorder Mania Neurological and symptomatic Autism spectrum Autism Asperger syndrome High-functioning autism PDD-NOS Savant syndrome Dementia AIDS dementia complex Alzheimer's disease Creutzfeldt–Jakob disease Frontotemporal dementia Huntington's disease Mild cognitive impairment Parkinson's disease Pick's disease Sundowning Vascular dementia Wandering Other Delirium Organic brain syndrome Post-concussion syndrome Neurotic , stress -related and somatoform Adjustment Adjustment disorder with depressed mood Anxiety Phobia Agoraphobia Social anxiety Social phobia Anthropophobia Specific social phobia Specific phobia Claustrophobia Other Generalized anxiety disorder OCD Panic attack Panic disorder Stress Acute stress reaction PTSD Dissociative Depersonalization disorder Dissociative identity disorder Fugue state Psychogenic amnesia Somatic symptom Body dysmorphic disorder Conversion disorder Ganser syndrome Globus pharyngis Psychogenic non-epileptic seizures False pregnancy Hypochondriasis Mass psychogenic illness Nosophobia Psychogenic pain Somatization disorder Physiological and physical behavior Eating Anorexia nervosa Bulimia nervosa Rumination syndrome Other specified feeding or eating disorder Nonorganic sleep Hypersomnia Insomnia Parasomnia Night terror Nightmare REM sleep behavior disorder Postnatal Postpartum depression Postpartum psychosis Sexual dysfunction Arousal Erectile dysfunction Female sexual arousal disorder Desire Hypersexuality Hypoactive sexual desire disorder Orgasm Anorgasmia Delayed ejaculation Premature ejaculation Sexual anhedonia Pain Nonorganic dyspareunia Nonorganic vaginismus Psychoactive substances, substance abuse and substance-related Drug overdose Intoxication Physical dependence Rebound effect Stimulant psychosis Substance dependence Withdrawal Schizophrenia , schizotypal and delusional Delusional Delusional disorder Folie à deux Psychosis and schizophrenia-like Brief reactive psychosis Schizoaffective disorder Schizophreniform disorder Schizophrenia Childhood schizophrenia Disorganized (hebephrenic) schizophrenia Paranoid schizophrenia Pseudoneurotic schizophrenia Simple-type schizophrenia Other Catatonia Symptoms and uncategorized Impulse control disorder Klüver–Bucy syndrome Psychomotor agitation Stereotypy Authority control GND : 4159331-5
Dyschondrosteosis - nephritis is characterized by the association of short stature due to mesomelic shortening of the limbs and Madelung deformity (see this term), with hereditary nephritis. Epidemiology It was originally described in male and female members from four generations of one large kindred. The females appeared to be more severely affected than the males, with a sex ratio (female to male) of 4:1. Clinical description The skeletal anomalies closely resembled those of Léri-Weill dyschondrosteosis (see this term). Genetic counseling The mode of transmission was reported as autosomal dominant.
Funderburk et al. (1976) described a kindred in which males and females in 4 generations appeared to have this combination. No male-to-male transmission was noted. Skel - Osteochondrodysplasia GU - Nephritis Growth - Short stature - Mesomelic dwarfism Neuro - Normal intelligence Facies - Normal Inheritance - Autosomal dominant vs. pseudoautosomal Joints - Limited elbow and wrist motion Limbs - Madelung deformity - Short forearm - Bowed radius - Bowed ulna - Dorsal dislocation of distal ulna - Short tibia Misc - Females more severely affected than males - 4:1 female-to-male ratio Head - Normocephaly ▲ Close
A rare ARX-related epileptic encephalopathy characterized by infantile onset of myoclonic epilepsy with generalized spasticity, severe global developmental delay, and moderate to profound intellectual disability. Obligate female carriers show subtle, generalized hyperreflexia. Late onset progressive spastic ataxia has also been reported.
Developmental and epileptic encephalopathy 1 (DEE1) is a seizure disorder characterized by a type of seizure known as infantile spasms. The spasms usually appear before the age of 1. Several types of spasms have been described, but the most commonly reported type involves bending at the waist and neck and extending the arms and legs (sometimes called a jackknife spasm). Each spasm lasts only seconds, but they occur in clusters several minutes long. Although individuals do not usually have spasms while they are sleeping, the spasms commonly occur just after awakening. Infantile spasms usually stop by age 5, but many children then develop other types of seizures that recur throughout their lives.
A monogenic disease with epilepsy characterized by developmental delay and infantile spasms in the first months of life, followed by chorea and generalized dystonia and progressing to quadriplegic dyskinesia, recurrent status dystonicus, intractable focal epilepsy and severe intellectual disability.
A rare genetic immune disease characterized by recurrent sinopulmonary infections and autoimmune enterocolopathy, manifesting as frequent episodes of intractable diarrhea with abdominal pain and fever, accompanied by eczematous rashes, due to deficits in components of innate and adaptive immunity. Immunologic abnormalities include IgG subclass deficiency, impaired antigen-induced lymphocyte proliferation, reduced cytokine production by CD8+ T lymphocytes, and decreased numbers of natural killer cells.
A rare complex hereditary spastic paraplegia characterized by neonatal to infantile onset of progressive spasticity in the lower limbs, hyperreflexia, tip-toe walking, pes equinus, and delayed motor developmental milestones. Kyphoscoliosis becomes evident in older patients, and most patients show atrophy of the lateral aspects of the tongue. Additional signs may include intellectual disability, language impairment, and moderate upper limb involvement.
A rare, genetic, inborn error of metabolism disorder characterized by global developmental delay, hypotonia, choreoathetosis, hypo-/alacrimia, and liver dysfunction which manifests with elevated liver transanimases and hepatocyte cytoplasmic storage material or vacuolization on liver biposy. Additional features reported include acquired microcephaly, hypo-/areflexia, seizures, peripheral neuropathy, intellectual and language/speech disability, additional ocular anomalies and EEG and brain imaging abnomalities.
A number sign (#) is used with this entry because congenital disorder of deglycosylation (CDDG) is caused by homozygous or compound heterozygous mutation in the NGLY1 gene (610661) on chromosome 3p24. Description Congenital disorder of deglycosylation is an autosomal recessive multisystem disorder characterized by global developmental delay, hypotonia, abnormal involuntary movements, and alacrima or poor tear production. Other common features include microcephaly, intractable seizures, abnormal eye movements, and evidence of liver dysfunction. Liver biopsy shows cytoplasmic accumulation of storage material in vacuoles (summary by Enns et al., 2014). For a discussion of the classification of congenital disorders of glycosylation, see CDG1A (212065).
Deficiency of N-glycanase 1 (NGLY1 deficiency) is a complex neurological syndrome in which there is a deficiency of an enzyme known as N-glycanase 1 (NGLY1).
NGLY1 -congenital disorder of deglycosylation ( NGLY1 -CDDG) is an inherited condition that affects many parts of the body. The severity of the signs and symptoms varies widely among people with the condition. Individuals with NGLY1 -CDDG typically develop features of the condition during infancy. They often have delayed development of speech and motor skills, such as sitting and walking, and weak muscle tone (hypotonia). Many affected individuals have movement abnormalities, such as uncontrolled movements of the limbs (choreoathetosis), and some develop seizures that are difficult to treat.
NGLY1 deficiency Other names NGLY1-congenital disorder of deglycosylation N-glycanase 1, the affected protein Specialty Medical genetics NGLY1 deficiency is a very rare genetic disorder caused by biallelic pathogenic variants in NGLY1 . It is an autosomal recessive disorder. Errors in deglycosylation are responsible for the symptoms of this condition. [1] Clinically, most affected individuals display developmental delay, lack of tears , elevated liver transaminases and a movement disorder. [2] NGLY1 deficiency is difficult to diagnose, and most individuals have been identified by exome sequencing. NGLY1 deficiency causes a dysfunction in the endoplasmic reticulum -associated degradation pathway. NGLY1 encodes an enzyme, N-glycanase 1 , that cleaves N-glycans . Without N-glycanase, N-glycosylated proteins that are misfolded in the endoplasmic reticulum cannot be degraded, and thus accumulate in the cytoplasm of cells. [3] [4] Contents 1 Signs and symptoms 2 Diagnosis 3 Treatment 4 Epidemiology 5 History 6 See also 7 References 8 External links Signs and symptoms [ edit ] Four common findings have been identified in a majority of patients: developmental delay or intellectual disability of varying degrees, lack of or greatly reduced tears , elevated liver transaminases, and a complex movement disorder. The elevated liver enzymes often resolve in childhood. In addition, approximately 50% of patients described with NGLY1 deficiency have seizures, which can vary in their difficulty to control.
However, because many of the clinical features overlap with those of other intellectual disability / developmental delay syndromes, a multigene panel or comprehensive genomic testing are typically used in lieu of single-gene testing. ... Alacrima & liver disease are not seen in disorders of creatine synthesis. Triple-A syndrome (OMIM 231550) AAAS AR Alacrima Mild dementia Cerebellar ataxia 13 Triple A syndrome does not feature choreoathetosis. Adrenal insufficiency is not a prominent feature of NGLY1-CDDG. Persons with Triple A syndrome may have anisocoria. Alacrima, achalasia, and mental retardation syndrome (AAMR) (OMIM 615510) GMPPA AR Alacrima ID Variable hypotonia Ataxia Spasticity Hearing impairment 14 AAMR: Does not feature choreoathetosis. ... Genetic disorders that may be associated with low HVA include mitochondrial disorders, glycine encephalopathy, Aicardi-Goutières syndrome, Rett syndrome (see MECP2 Disorders), myotonic dystrophy type 1, and vanishing white matter disease (see Childhood Ataxia with Central Nervous System Hypomyelination/Vanishing White Matter). 11. ... Genetic disorders that may be associated with low HVA and 5-HIAA include mitochondrial disease, Niemann-Pick disease type C, Alexander disease, glycine encephalopathy, pontocerebellar hypoplasia type 2 (see TSEN54 -Related Pontocerebellar Hypoplasia), Rett syndrome (see MECP2 Disorders), Smith-Lemli-Opitz syndrome, urea cycle disorders [Molero-Luis et al 2013, Ng et al 2015]. 13.
A rare genetic neurological disorder characterized by infantile to childhood onset of global developmental delay, hypotonia, seizures, growth delay, and intellectual disability. Additional variable features include strabismus, cortical visual impairment, nystagmus, movement disorder (such as dystonia, ataxia, or chorea), or mild dysmorphic features, among others.
A number sign (#) is used with this entry because of evidence that autosomal dominant mental retardation-42 (MRD42) is caused by heterozygous mutation in the GNB1 gene (139380) on chromosome 1p36. Description Autosomal dominant mental retardation-42 is a neurodevelopmental disorder characterized by global developmental delay and intellectual disability. More variable features include hypotonia, often later associated with limb hypertonia, seizures of various types, and poor overall growth. Strabismus, cortical visual impairment, and autistic features may also be present (summary by Petrovski et al., 2016). Clinical Features Petrovski et al. (2016) reported 13 unrelated individuals, ranging in age from 13 months to 20 years, with global developmental delay.
A rare genetic disease characterized by early-onset severe obesity due to mutations in single genes acting on the development and function of the hypothalamus or the leptin-melanocortin pathway, leading to disruption of energy homeostasis and endocrine dysfunction. Patients present with a body mass index over three standard deviations above normal at less than five years of age, accompanied by a variety of signs and symptoms according to the mutated gene, including hyperphagia, insulin resistance, reduced basal metabolic rate, or hypogonadism, among others.
A rare congenital myopathy characterized by early onset of severe muscular weakness, respiratory distress due to diaphragmatic paralysis, dysphagia and areflexia, joint contractures, and scoliosis. Decreased fetal movements are seen in some individuals. Muscle biopsy may show a combination of dystrophic and myopathic features. The clinical course is variable, with some patients becoming ventilator-dependent and never achieving ambulation.
A number sign (#) is used with this entry because early-onset myopathy, areflexia, respiratory distress, and dysphagia (EMARDD) is caused by homozygous or compound heterozygous mutation in the MEGF10 gene (612453) on chromosome 5q23. Description EMARDD is a congenital myopathy characterized by proximal and generalized muscle weakness, respiratory difficulties, joint contractures, and scoliosis. More variable features include cleft palate and feeding difficulties. There is variable severity: some patients become ventilator-dependent, never achieve walking, and die in childhood, whereas others have a longer and more favorable course (summary by Logan et al., 2011 and Boyden et al., 2012). Clinical Features Hartley et al. (2007) reported 2 unrelated but consanguineous families from Qatar and Sri Lanka, respectively, in which 2 sibs in each family had a severe congenital myopathy particularly affecting the diaphragm.
A rare genetic neurological disorder characterized by postnatal microcephaly, hypotonia during infancy followed in most cases by progressive spasticity mainly affecting the lower limbs, and spastic diplegia or paraplegia, intellectual disability, delayed or absent speech, and dysarthria. Seizures and mildly dysmorphic features have been described in some patients.
A number sign (#) is used with this entry because of evidence that autosomal recessive mental retardation-49 (MRT49) is caused by homozygous mutation in the GPT2 gene (138210) on chromosome 16q11. Clinical Features Celis et al. (2015) reported a distantly consanguineous Mizrahi Jewish family with 3 affected sibs (2 boys and 1 girl) who had developmental encephalopathy characterized by rapid onset of failure to thrive and microcephaly as well as profoundly delayed development. All 3 children were nonverbal. The oldest child, a 15-year-old boy, developed generalized tonic-clonic seizures at 10 years of age. His 14-year-old affected sister developed seizures at the age of 14. Both older sibs were hypotonic. The youngest affected sib, a 4-year-old boy, was hypertonic and exhibited fisting.