A rare neurologic disease characterized by axonal sensorimotor neuropathy, progressive optic atrophy, cognitive deficit, bulbar dysfunction, seizures, and early hypotonia and feeding difficulties. Additional possible features include dystonia, scoliosis, joint contractures, ocular anomalies, and urogenital anomalies. Brain MRI reveals variable degrees of cerebral atrophy. The disease is fatal in childhood due to respiratory failure.
A rare genetic endocrine disease characterized by intrauterine growth restriction, failure of an adolescent growth spurt with proportional adult short stature, insulin resistance, and early adulthood-onset diabetes. Minimal subluxation of the fifth metacarpal-phalangeal joint has been reported, while metaphyseal dysplasia is absent. Testicular volume is low, but fertility is normal. There is no evidence of primary adrenal insufficiency.
A rare acute encephalopathy with inflammation-mediated status epilepticus characterized by infancy-onset of refractory unilateral, mainly clonic status epilepticus during or shortly after a febrile episode without evidence of central nervous system infection, followed by permanent or transient hemiplegia with a minimum duration of one week. The majority of children develop pharmaco-resistant epilepsy a few months later. Brain imaging shows edematous swelling of the affected hemisphere at the time of the initial status, followed by hemiatrophy that does not correlate with any vascular territory.
A rare genetic disease characterized by global developmental delay with language and cognition deficiencies, behavioral problems, osteopenia, joint laxity, skin defects consisting of hyperkeratosis and sweat gland and melanocyte abnormalities with hypopigmented areas, and abnormal hair structure. Mild facial dysmorphism (prominent forehead, thick eyebrows, epicanthal folds, broad nasal bridge, long philtrum, and micrognathia), abnormalities of the teeth, and skeletal and cardiac anomalies have also been described.
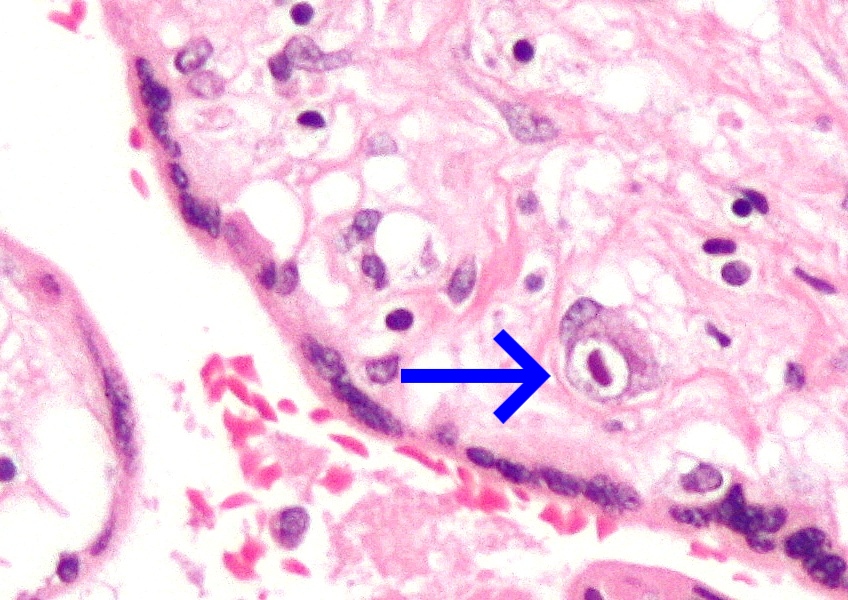
A fetopathy that is likely to occur when a cytomegalovirus (CMV) infected pregnant woman transmits the virus in utero . Children born with congenital CMV infection may present with hepatomegaly, splenomegaly, jaundice, pneumonitis, fetal growth retardation, petechiae, purpura, and thrombocytopenia. Congenital CMV infection can equally result in major neurological sequelae, including microcephaly, intracranial calcifications, sensorineural hearing loss, chorioretinitis, intellectual and motor disabilities, and seizure disorders. CMV disease sequelae caused by a primary infection are usually more severe than those caused by the reactivation of a latent infection.
Contents 1 Presentation 2 Prevention 2.1 Childcare 2.2 CMV Testing and Diagnosis 3 Epidemiology 4 References 5 External links Presentation [ edit ] For infants who are infected by their mothers before birth, two potential adverse scenarios exist: Generalized infection may occur in the infant, and can cause complications such as low birth weight , microcephaly , seizures , petechial rash similar to the "blueberry muffin" rash of congenital rubella syndrome , and moderate hepatosplenomegaly (with jaundice ). ... A delay in general speech and language development is more common in children with CMV. [8] Children with symptomatic CMV have been found to have a greater incidence of long-term neurological and neurodevelopmental complications than children with fetal alcohol syndrome or down syndrome. [4] Congenital cytomegalovirus infection can be an important cause of intraventricular hemorrhage and neonatal encephalopathy . [9] Prevention [ edit ] Recommendations for pregnant women with regard to CMV infection: [ citation needed ] Throughout the pregnancy, practice good personal hygiene, especially handwashing with soap and water, after contact with diapers or oral secretions (particularly with a child who is in day care). ... HCMV is the most common cause of congenital infection in humans and intrauterine primary infections are more common than other well-known infections and syndromes, including Down Syndrome, Fetal Alcohol Syndrome, Spina Bifida, and Pediatric HIV/AIDS. [ citation needed ] References [ edit ] ^ a b c d Dobbie, Allison M. (2017). ... "Breast milk as a source for acquisition of cytomegalovirus (HCMV) in a premature infant with sepsis syndrome: detection by real-time PCR". J. ... External links [ edit ] Cytomegalovirus (CMV) —NHS Choices CMV: Congenital CMV Infection —CDC Classification D ICD - 10 : P35.1 ICD - 9-CM : 771.1 External resources MedlinePlus : 001343 eMedicine : article/963090 v t e Vertically transmitted infections Gestational Viruses Congenital rubella syndrome Congenital cytomegalovirus infection Neonatal herpes simplex Hepatitis B Congenital varicella syndrome HIV Fifth disease Bacteria Congenital syphilis Other Toxoplasmosis transplacental TORCH complex During birth transcervical Candidiasis Gonorrhea Listeriosis Late pregnancy Listeriosis Congenital cytomegalovirus infection By breastfeeding Breastfeeding Tuberculosis HIV
A rare, genetic, parenchymal hepatic disease characterized by acute liver failure, that occurs in the first year of life, which manifests with failure to thrive, hypotonia, moderate global developmental delay, seizures, abnormal liver function tests, microcytic anemia and elevated serum lactate. Other associated features include hepatosteatosis and fibrosis, abnormal brain morphology, and renal tubulopathy. Minor illness exacerbates deterioration of liver failure.
A number sign (#) is used with this entry because of evidence that infantile liver failure syndrome-1 (ILFS1) is caused by homozygous mutation in the LARS gene (151350) on chromosome 5q32. ... Genetic Heterogeneity of Infantile Liver Failure Syndrome See also ILFS2 (616483), caused by mutation in the NBAS gene (608025) on chromosome 2p24.
Partial monosomy of the long arm of chromosome 9 characterized by intellectual disability, developmental delay with pronounced speech delay, short stature, and muscular hypotonia. Common craniofacial dysmorphic features consist of microcephaly, prominent forehead, round face, arched eyebrows, upslanting palpebral fissures, strabismus, short nose, and thin upper lip. Other clinical findings include epilepsy, ataxia, unspecific brain MRI findings, early-onset primary dystonia, nail dysplasia, and bone malformations, in particular patellar abnormalities, epistaxis, and cutaneous-mucous telangiectasias.
A rare disorder of manganese transport characterized by progressive movement disorder and elevated blood manganese levels. Patients present in infancy or early childhood with loss of motor milestones, rapidly progressive dystonia, spasticity, bulbar dysfunction, and parkinsonism, resulting in loss of independent ambulation. Cognition may be impaired but is generally better preserved than motor function. Additional manifestations include abnormal head growth and skull deformities. Brain MRI shows abnormalities of the basal ganglia, variably also of other brain regions.
A number sign (#) is used with this entry because of evidence that hypermanganesemia with dystonia-2 (HMNDYT2) is caused by homozygous mutation in the SLC39A14 gene (608736) on chromosome 8p21. Description Hypermanganesemia with dystonia-2 is an autosomal recessive neurodegenerative disorder characterized predominantly by loss of motor milestones in the first years of life. Affected individuals then develop rapidly progressive abnormal movements, including dystonia, spasticity, bulbar dysfunction, and variable features of parkinsonism, causing loss of ambulation. Cognition may be impaired, but is better preserved than motor function. The disorder results from abnormal accumulation of manganese (Mn), which is toxic to neurons.
A rare genetic disease characterized by abnormalities in renal ion transport, ectodermal gland homeostasis, and epidermal integrity, resulting in generalized hypohidrosis, heat intolerance, salt-losing nephropathy, electrolyte imbalance, lacrimal gland dysfunction, ichthyosis, and xerostomia. Development of nephrolithiasis and severe enamel wear have also been described. Laboratory findings include hypermagnesemia, hypokalemia, hypercalcemia, and hypocalciuria.
A number sign (#) is used with this entry because of evidence that HELIX syndrome (HELIX) is caused by homozygous mutation in the CLDN10 gene (617579) on chromosome 13q32. ... Hadj-Rabia et al. (2018) reported 6 patients from 2 unrelated consanguineous families with hypohidrosis, electrolyte imbalance, lacrimal gland dysfunction, ichthyosis, and xerostomia, which the authors designated 'HELIX syndrome.' Dental examination showed severe enamel wear; panoramic dental x-rays of patients from both families showed that enamel formed but eroded quickly after tooth eruption. ... Hadj-Rabia et al. (2018) suggested that the renal NaCl wasting syndrome observed in these patients was primarily due to electrolyte transport disturbances in the thick ascending limb of the loop of Henle.
This syndrome is characterised by childhood-onset progressive ataxia and cerebellar atrophy. ... Clinical description Exercise intolerance with elevated lactate levels and mild intellectual deficit may also be present. Etiology The syndrome is caused by ubiquinone deficiency. ... This gene is already known to play a role in ubiquinone biosynthesis in yeast. Genetic counseling The syndrome is transmitted as an autosomal recessive trait.
A number sign (#) is used with this entry because primary coenzyme Q10 deficiency-4 (COQ10D4), also known as autosomal recessive spinocerebellar ataxia-9 (SCAR9), is caused by homozygous or compound heterozygous mutation in the ADCK3 gene (COQ8A; 606980) on chromosome 1q42. Description Primary coenzyme Q10 deficiency-4 is an autosomal recessive disorder characterized by childhood-onset of cerebellar ataxia and exercise intolerance. Some affected individuals develop seizures and have mild mental impairment, indicating variable severity. Oral coenzyme Q10 supplementation does not result in significant improvement of neurologic symptoms (summary by Mollet et al., 2008 and Lagier-Tourenne et al., 2008). For a general phenotypic description and a discussion of genetic heterogeneity of primary coenzyme Q10 deficiency, see COQ10D1 (607426).
A rare mitochondrial disease characterized by neonatal onset of severe cardiac and/or neurologic signs and symptoms mostly associated with a fatal outcome in the neonatal period or in infancy, although a milder phenotype with later onset and slowly progressive neurologic deterioration has also been reported. Clinical manifestations are variable and include respiratory insufficiency, hypotonia, cardiomyopathy, and seizures. Serum lactate is elevated in most cases. Brain imaging may show cerebellar atrophy or hypoplasia.
A number sign (#) is used with this entry because of evidence that primary coenzyme Q10 deficiency-7 (COQ10D7) is caused by homozygous or compound heterozygous mutation in the COQ4 gene (612898) on chromosome 9q34. Description Primary coenzyme Q10 deficiency-7 is an autosomal recessive disorder resulting from mitochondrial dysfunction. Most patients have onset of severe cardiac or neurologic symptoms soon after birth, usually resulting in death. Rare patients may have later onset with a more protracted course. Tissue samples from affected individuals show decreased levels of coenzyme Q10 (CoQ10) (summary by Brea-Calvo et al., 2015). For a general phenotypic description and a discussion of genetic heterogeneity of primary coenzyme Q10 deficiency, see COQ10D1 (607426).
A number sign (#) is used with this entry because of evidence that lateral meningocele syndrome (LMNS) is caused by heterozygous mutation in the NOTCH3 gene (600276) on chromosome 19p13. ... Merle et al. (1979) suggested a general dysplastic syndrome rather than a local disease. Philip et al. (1995) reported a 19-year-old boy with facial dysmorphism, skeletal anomalies, and multiple lateral meningoceles. ... Chen et al. (2005) suggested that lateral meningocele syndrome may be a connective tissue disorder. ... Avela et al. (2011) suggested that the diagnosis in this patient was consistent with Hajdu-Cheney syndrome (HJCYS; 102500). However, Gripp (2011) concluded that the patient reported by Avela et al. (2011) had lateral meningocele syndrome, although she noted that the 2 disorders have overlapping features and may either be allelic or caused by mutations in genes in the same pathway. ... Alves et al. (2013) emphasized the connective tissue defects often observed in lateral meningocele syndrome. Gripp et al. (2015) reported a boy with lateral meningocele syndrome who developed acute back pain, urinary incontinence, worsening stool incontinence, and abnormal gait around 5 years of age.
Craniolenticulosutural dysplasia (CLSD), also known as Boyadjiev-Jabs syndrome, is characterized by the specific association of large and late-closing fontanels, hypertelorism, early-onset cataract and mild generalized skeletal dysplasia.
Clinical Features Boyadjiev et al. (2003) suggested the designation craniolenticulosutural dysplasia (CLSD) for a dysmorphic syndrome in 5 males and 1 female in an inbred Saudi Arabian family.
It is very rare that globus sensation presenting with no other symptoms has a sinister cause and therefore endoscopy is not recommended in this case. [9] [10] Differential diagnosis [ edit ] Differential diagnosis must be made from Eagle syndrome which uses the patient's description of "something caught in my throat" as a diagnostic tool. Eagle syndrome is an elongation of the styloid process causing irritation to nerves and muscles in the region resulting in a number of unusual symptoms. [ citation needed ] Management [ edit ] Reassurance of the patient is recommendable when no cause can be found. ... Classification D ICD - 10 : F45.8 ICD - 9-CM : 300.11 MeSH : D003291 DiseasesDB : 31559 External resources Patient UK : Globus pharyngis v t e Mental and behavioral disorders Adult personality and behavior Gender dysphoria Ego-dystonic sexual orientation Paraphilia Fetishism Voyeurism Sexual maturation disorder Sexual relationship disorder Other Factitious disorder Munchausen syndrome Intermittent explosive disorder Dermatillomania Kleptomania Pyromania Trichotillomania Personality disorder Childhood and learning Emotional and behavioral ADHD Conduct disorder ODD Emotional and behavioral disorders Separation anxiety disorder Movement disorders Stereotypic Social functioning DAD RAD Selective mutism Speech Stuttering Cluttering Tic disorder Tourette syndrome Intellectual disability X-linked intellectual disability Lujan–Fryns syndrome Psychological development ( developmental disabilities ) Pervasive Specific Mood (affective) Bipolar Bipolar I Bipolar II Bipolar NOS Cyclothymia Depression Atypical depression Dysthymia Major depressive disorder Melancholic depression Seasonal affective disorder Mania Neurological and symptomatic Autism spectrum Autism Asperger syndrome High-functioning autism PDD-NOS Savant syndrome Dementia AIDS dementia complex Alzheimer's disease Creutzfeldt–Jakob disease Frontotemporal dementia Huntington's disease Mild cognitive impairment Parkinson's disease Pick's disease Sundowning Vascular dementia Wandering Other Delirium Organic brain syndrome Post-concussion syndrome Neurotic , stress -related and somatoform Adjustment Adjustment disorder with depressed mood Anxiety Phobia Agoraphobia Social anxiety Social phobia Anthropophobia Specific social phobia Specific phobia Claustrophobia Other Generalized anxiety disorder OCD Panic attack Panic disorder Stress Acute stress reaction PTSD Dissociative Depersonalization disorder Dissociative identity disorder Fugue state Psychogenic amnesia Somatic symptom Body dysmorphic disorder Conversion disorder Ganser syndrome Globus pharyngis Psychogenic non-epileptic seizures False pregnancy Hypochondriasis Mass psychogenic illness Nosophobia Psychogenic pain Somatization disorder Physiological and physical behavior Eating Anorexia nervosa Bulimia nervosa Rumination syndrome Other specified feeding or eating disorder Nonorganic sleep Hypersomnia Insomnia Parasomnia Night terror Nightmare REM sleep behavior disorder Postnatal Postpartum depression Postpartum psychosis Sexual dysfunction Arousal Erectile dysfunction Female sexual arousal disorder Desire Hypersexuality Hypoactive sexual desire disorder Orgasm Anorgasmia Delayed ejaculation Premature ejaculation Sexual anhedonia Pain Nonorganic dyspareunia Nonorganic vaginismus Psychoactive substances, substance abuse and substance-related Drug overdose Intoxication Physical dependence Rebound effect Stimulant psychosis Substance dependence Withdrawal Schizophrenia , schizotypal and delusional Delusional Delusional disorder Folie à deux Psychosis and schizophrenia-like Brief reactive psychosis Schizoaffective disorder Schizophreniform disorder Schizophrenia Childhood schizophrenia Disorganized (hebephrenic) schizophrenia Paranoid schizophrenia Pseudoneurotic schizophrenia Simple-type schizophrenia Other Catatonia Symptoms and uncategorized Impulse control disorder Klüver–Bucy syndrome Psychomotor agitation Stereotypy
In some cases these substance induced psychiatric disorders can persist long after detoxification, such as prolonged psychosis or depression after amphetamine or cocaine abuse. A protracted withdrawal syndrome can also occur with symptoms persisting for months after cessation of use. ... By 1988, the DSM-IV defines substance dependence as "a syndrome involving compulsive use, with or without tolerance and withdrawal"; whereas substance abuse is "problematic use without compulsive use, significant tolerance, or withdrawal."