This article is about the cutaneous condition Bazex syndrome. For the inherited condition Bazex syndrome, see Bazex–Dupré–Christol syndrome . Paraneoplastic acrokeratosis Other names Acrokeratosis paraneoplastica of Bazex and Acrokeratosis neoplastica ) [1] Specialty Dermatology Paraneoplastic acrokeratosis , or Bazex syndrome is a cutaneous condition characterized by psoriasiform changes of hands, feet, ears, and nose, with involvement of the nails and periungual tissues being characteristic and indistinguishable from psoriatic nails .
A number sign (#) is used with this entry because of evidence that coloboma of the optic nerve and morning glory disc anomaly are each caused by mutation in the PAX6 gene (607108). One such patient has been reported for each of the phenotypes. Clinical Features Congenital coloboma of the optic nerve is often associated with serious detachment of the macula. Savell and Cook (1976) observed 15 affected persons in 1 kindred. In 21 of the 30 eyes, present or past detachment of the retina was found. The coloboma was bilateral in all. It appeared as enlargement of the physiologic cup with severely affected eyes showing huge cavities at the site of the disc. A variable amount of glial tissue was present in the coloboma. No male-to-male transmission was observed.
MGDA can be associated with Moyamoya disease, PHACES syndrome, Aicardi syndrome, neurofibromatosis type 2, Arnold-Chiari malformation type I, CHARGE syndrome, Poland syndrome, among others. ... Etiology The exact etiology is not fully understood, but the syndrome is related to poor development of the posterior sclera and lamina cribrosa during gestation.
The development of multiple cylindromas can be hereditary and is inherited in an autosomal dominant manner; this condition is called CYLD cutaneous syndrome . Individuals with the inherited form begin to develop many, rounded nodules of various size shortly after puberty.
Dysequilibrium syndrome (DES) is a non-progressive cerebellar disorder characterized by ataxia associated with an intellectual disability, delayed ambulation and cerebellar hypoplasia.
A number sign (#) is used with this entry because of evidence that cerebellar ataxia, mental retardation, and dysequilibrium syndrome-2 (CAMRQ2) is caused by homozygous mutation in the WDR81 gene (614218) on chromosome 17p. Description Cerebellar ataxia, mental retardation, and dysequilibrium syndrome (CAMRQ) is a genetically heterogeneous disorder characterized by congenital cerebellar ataxia and mental retardation (summary by Gulsuner et al., 2011). ... He noted that the affected individuals demonstrated diagonal walking seen in many animals, such as dogs, horses, and chimpanzees. Tan (2006) suggested that this syndrome may represent a live model for human evolution from quadrupedal to bipedal gait. ... Garcias and Martino Roth (2007) reported 4 Brazilian sibs, 2 males and 2 females, born of consanguineous parents, with a clinically homogeneous syndrome in which the predominant characteristic was a quadrupedal gait. ... Molecular Genetics In affected members of a consanguineous Turkish family with autosomal recessive cerebellar ataxia, mental retardation, and dysequilibrium syndrome-2 (Turkmen et al., 2006), Gulsuner et al. (2011) identified a homozygous mutation in the WDR81 gene (P856L; 614218.0001).
VLDLR-associated cerebellar hypoplasia Other names Dysequilibrium syndrome , DES ; Nonprogressive cerebellar disorder with mental retardation ) VLDLR-associated cerebellar hypoplasia ( VLDLRCH ) is a rare autosomal recessive condition caused by a disruption of the VLDLR gene. [1] [2] First described as a form of cerebral palsy in the 1970s, [3] it is associated with parental consanguinity and is found in secluded communities, with a number of cases described in Hutterite families. [4] References [ edit ] ^ Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, Davey K, Chudley AE, Scott JN, McLeod DR, Parboosingh JS (2005). ... "Identification of a nonsense mutation in the very low-density lipoprotein receptor gene (VLDLR) in an Iranian family with dysequilibrium syndrome" . Eur. J. Hum. Genet . 16 (2): 270–3. doi : 10.1038/sj.ejhg.5201967 . PMID 18043714 . ^ Sanner, G. The dysequilibrium syndrome: a genetic study. Neuropaediatrie 4: 403-413, 1973 ^ Schurig V, Orman AV, Bowen P (1981).
A number sign (#) is used with this entry because cerebellar ataxia, mental retardation, and dysequilibrium syndrome (CAMRQ1) is caused by homozygous mutation in the VLDLR gene (192977), which encodes the very low density lipoprotein receptor, on chromosome 9p24. ... Baraitser (1993) referred to a pair of sibs with a clinical picture that would fit with a diagnosis of 'ataxic cerebral palsy'; progression of the ataxia was so slight that the correct diagnosis of carbohydrate-deficient glycoprotein syndrome (212065) was missed. Mapping Boycott et al. (2005) used an identity-by-descent mapping approach in 8 patients from 3 interrelated Hutterite families to localize the gene for this syndrome to 9p24. ... This condition appeared to represent the first example of a malformation syndrome due to a defect in a human lipoprotein receptor and the second human disease associated with a reelin pathway defect. The other is a syndrome of autosomal recessive lissencephaly with cerebellar hypoplasia (257320) due to mutation in the RELN gene. ... History In 8 affected children with dysequilibrium syndrome, defined as an autosomal recessive form of cerebral palsy, Gustavson et al. (1977) found low serum dopamine-beta-hydroxylase activity.
Description Cerebellar ataxia, mental retardation, and dysequilibrium syndrome (CAMRQ) is a genetically heterogeneous disorder characterized by congenital cerebellar ataxia and mental retardation (summary by Gulsuner et al., 2011). ... Molecular Genetics In 3 affected members of a consanguineous Turkish family with cerebellar ataxia, mental retardation, and dysequilibrium syndrome (CAMRQ4), Onat et al. (2013) identified a homozygous mutation in the ATP8A2 gene (I376M; 605870.0001).
Description Cerebellar ataxia, mental retardation, and dysequilibrium syndrome (CAMRQ) is a genetically heterogeneous disorder characterized by congenital cerebellar ataxia and mental retardation (summary by Gulsuner et al., 2011). ... By homozygosity mapping followed by exon enrichment and next-generation sequencing in 136 consanguineous families (over 90% Iranian and less than 10% Turkish or Arabic) segregating syndromic or nonsyndromic forms of autosomal recessive intellectual disability, Najmabadi et al. (2011) identified a missense mutation in the CA8 gene (114815.0002) as the cause of CAMRQ3 in a family (M107) in which first-cousin parents had 2 healthy and 4 affected children.
Hamann et al. (1992) concluded that this represented a new syndrome, although the possibility of a contiguous gene syndrome was considered. Skel - Multiple exostoses Neuro - Spastic tetraparesis Inheritance - Autosomal dominant - possibly a contiguous gene syndrome ▲ Close
A rare genetic disease characterized by juvenile-onset insulin-dependent diabetes mellitus associated with central and peripheral nervous system abnormalities with variable onset between infancy and adolescence. Neurological manifestations include combined cerebellar and afferent ataxia, sensorineural hearing loss, pyramidal tract signs, and demyelinating sensorimotor peripheral neuropathy. Hypothyroidism has been reported in some patients. Brain imaging may show generalized cerebral atrophy.
A number sign (#) is used with this entry because of evidence that combined cerebellar and peripheral ataxia with hearing loss and diabetes mellitus (ACPHD) is caused by homozygous mutation in the DNAJC3 gene (601184) on chromosome 13q32. One such family has been reported. Clinical Features Synofzik et al. (2014) reported 3 adult sibs, from a 'likely consanguineous' (Synofzik, 2015) Turkish family, with juvenile-onset insulin-dependent diabetes mellitus and central and peripheral nervous system abnormalities. The neurologic features included combined cerebellar and afferent ataxia, mild upper motor neuron damage, demyelinating sensorimotor peripheral neuropathy, and sensorineural hearing loss. All 3 patients had onset of diabetes between 15 and 18 years of age. The age at onset of the neurologic features ranged widely: 1 sib had onset of hearing loss and gait disturbances at age 6, another had onset of these symptoms in the teenage years, and the third had onset of hearing loss at age 27 and gait disturbances at age 34. The patients also had an 'isolated cognitive deficit in backward calculation of serial sevens.'
A rare genetic neurological disorder characterized by the association of congenital spastic paraplegia with global developmental delay and intellectual disability, ophthalmologic abnormalities (including nystagmus, reduced visual acuity, or hypermetropia), and obesity. Additional manifestations are brachyplagiocephaly and dysmorphic facial features. Brain imaging may show dilated ventricles, abnormal myelination, and mild generalized atrophy. Homozygous loss-of-function variants of KIDINS220 associated with a fetal lethal phenotype with ventriculomegaly and limb contractures have been reported.
Clinical Features Josifova et al. (2016) reported 3 unrelated children with a similar intellectual disability syndrome. Prenatal ultrasound of all 3 showed dilated lateral ventricles.
A rare genetic neurological disorder characterized by infantile or childhood onset of recurrent acute encephalopathic episodes with cerebellar and extrapyramidal involvement following febrile illnesses. During the episodes, patients typically show sudden onset of truncal ataxia, occasionally accompanied by lethargy and impairment of speech, as well as choreic and athetoid movements, seizures, loss of deep tendon reflexes, and presence of pathological reflexes. Episodes last from day to weeks and may leave residual symptoms such as speech impairment and poor coordination. There have been no further descriptions in the literature since 1983.
Clinical Features Neuhauser et al. (1983) described 2 unrelated families in which a total of 5 members had recurrent encephalopathy affecting cerebellar and extrapyramidal structures. Affected individuals had onset in infancy or early childhood of acute encephalopathic episodes following presumably viral infections. The episodes were characterized by lethargy, poor coordination, tremor, involuntary movements, and loss of coordination and voluntary motor skills. All psychomotor abilities were fully regained after these initial episodes; however, the attacks were recurrent during childhood, and all patients later developed residual neurologic impairment, including hypotonia, areflexia, extensor plantar responses, choreoathetoid movements, ataxia, intention tremor, and variable dysarthria. Inheritance The transmission pattern in the families reported by Neuhauser et al. (1983) was consistent with autosomal dominant inheritance.
A rare genetic neurological disorder characterized by infantile hypotonia, congenital ophthalmic anomalies (including strabismus, esotropia, nystagmus, and central visual impairment), global developmental delay and intellectual disability, behavioral abnormalities, and movement disorder (such as dystonia, chorea, hyperkinesia, stereotypies). Mild facial dysmorphism and skeletal deformities have also been reported. EEG testing shows marked abnormalities in the absence of overt epileptic seizures.
A number sign (#) is used with this entry because of evidence that Baker-Gordon syndrome (BAGOS) is caused by heterozygous mutation in the SYT1 gene (185605) on chromosome 12q21. Description Baker-Gordon syndrome (BAGOS) is a neurodevelopmental disorder characterized by infantile hypotonia, ophthalmic abnormalities, moderate to profound global developmental delay, poor or absent speech, behavioral abnormalities, hyperkinetic movements, and EEG abnormalities in the absence of overt seizures (summary by Baker et al., 2018).
An extremely rare, autosomal recessive, hereditary cerebellar ataxia disorder characterized by early onset of progressive, mild to moderate gait and limb ataxia, moderate to severe dysarthria, and nystagmus or saccadic pursuit, frequently associated with epilepsy, moderate intellectual disability, delayed speech acquisition, and hyporeflexia in the upper extremities. Hyperreflexia in the lower extremities may also be associated.
A number sign (#) is used with this entry because of evidence that autosomal recessive spinocerebellar ataxia-15 (SCAR15) is caused by homozygous mutation in the KIAA0226 gene (RUBCN; 613516) on chromosome 3q29. One such family has been reported. Clinical Features Assoum et al. (2010) reported a consanguineous Saudi Arabian family in which 3 sisters had onset of cerebellar ataxia in early childhood. All showed delayed motor development with delayed walking. Two sisters had a more severe form of the disorder, with an unsteady gait apparent since learning to walk, whereas the third developed unsteady gait around age 7 years. Other features included dysarthria, upper limb involvement, abnormal eye movements, and hyporeflexia. Two patients had increased reflexes in the lower limbs. At age 7 months the 2 sisters who were more severely affected developed epilepsy, which was responsive to treatment with no relapse in either girl since age 3 years; both subsequently showed moderate mental retardation.
A rare genetic systemic or rheumatologic disease characterized by neonatal or infantile onset of enterocolitis (which resolves with age), periodic fever, and episodes of severe systemic inflammation, which may be precipitated by infections, stress, or fatigue. Signs and symptoms include splenomegaly, urticaria-like rashes, arthralgia, and myalgia. Associated laboratory findings are elevated inflammatory markers (such as ferritin, C-reactive protein), pancytopenia, and elevated transaminases. If left untreated, flares can progress to coagulopathy, organ failure, and death.
Clinical Features Romberg et al. (2014) reported a father and his 2 sons with an autoinflammatory syndrome characterized by neonatal-onset enterocolitis, periodic fever, and fatal or near-fatal episodes of autoinflammation. ... Canna et al. (2014) reported a 7-year-old girl of European descent with a recurrent autoinflammatory syndrome. She presented at age 2 months with failure to thrive associated with anemia and increased serum ferritin.
Beta-thalassemia - X-linked thrombocytopenia is a form of beta-thalassemia (see this term) characterized by splenomegaly and petechiae, moderate thrombocytopenia, prolonged bleeding time due to platelet dysfunction, reticulocytosis and mild beta-thalassemia. Epidemiology Prevalence of this form is not known. Etiology The disorder is not associated with mutations in the HBB gene (11p15.5), but with mutations in the gene encoding GATA-binding protein-1 ( GATA1 ; Xp11.23) that result in reduced expression of the beta-globin genes. Genetic counseling Transmission is X-linked.
Tubman et al. (2007) identified an R216Q substitution in affected members of a family with a mild bleeding disorder, thrombocytopenia, and large agranular platelets characteristic of the so-called 'gray platelet syndrome.' In a letter, Balduini et al. (2007) stated that the family reported by Tubman et al. (2007) had a phenotype consistent with XLTT and that the classification as 'X-linked gray platelet syndrome' is a misnomer risking confusion in the literature.
Commonly, however, macrodontia is associated with genetic syndromes such as otodental syndrome , insulin-resistant diabetes , facial hemihyperplasia , KGB syndrome , Ekman-Westborg-Julian syndrome, and 47 XYY syndrome . ... Thus, macrodontia is exhibited due to the overexpression of GH through a form of gigantism . Another key syndrome which results in the expression of macrodontia is KBG syndrome . ... Other patients may have a disease called Rabson-Mendenhall syndrome which predisposes to generalized macrodontia. ... Because the nature of macrodontia is mostly due to genetic syndromes, the specialist will likely recommend that a patient visit a cosmetic dentist. ... "Mutations in ANKRD11 Cause KBG Syndrome, Characterized by Intellectual Disability, Skeletal Malformations, and Macrodontia" .
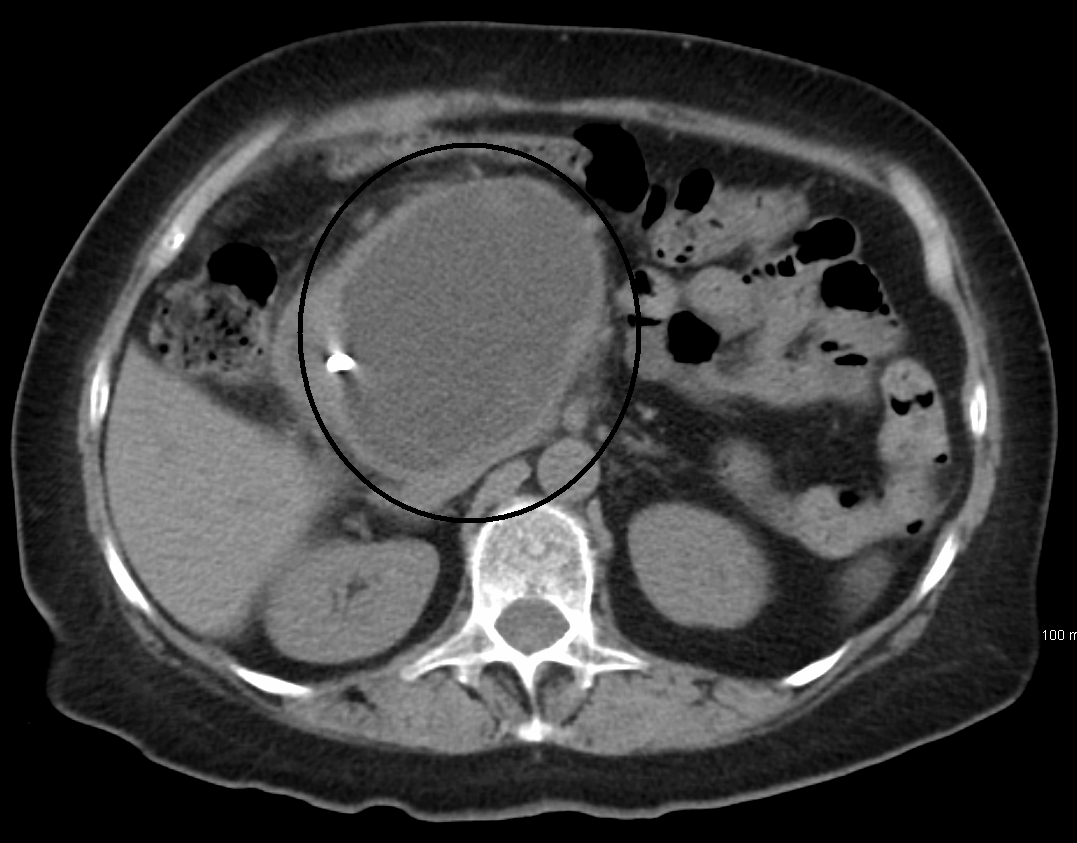
Overview Median arcuate ligament syndrome (MALS) occurs when the arc-shaped band of tissue in the chest area (median arcuate ligament) presses on the artery that sends blood to the upper abdomen. ... Other names for MALS are: Celiac artery compression Celiac axis syndrome Dunbar syndrome Treatment involves surgery to release (decompress) the ligament and restore blood flow through the artery. ... However, those with median arcuate ligament syndrome (MALS) can have long-term (chronic) stomach pain. ... Risk factors Because the cause of MALS is poorly understood, the risk factors for the syndrome are unclear. MALS has been seen in children, even twins, which might mean genetics plays a role. ... List your questions from most to least important in case time runs out. For median arcuate ligament syndrome, some basic questions to ask your provider include: What is likely causing my symptoms or condition?
External links [ edit ] Classification D ICD - 10 : K72.0 External resources MedlinePlus : 000214 Scholia has a topic profile for Ischemic hepatitis . v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
Idiopathic CD4+ lymphocytopenia Other names Immunodeficiency 13 Idiopathic CD4+ lymphocytopenia is inherited via autosomal dominant manner [1] Specialty Immunology Idiopathic CD4+ lymphocytopenia ( ICL ) is a rare medical syndrome in which the body has too few CD4 + T lymphocytes , which are a kind of white blood cell . [2] ICL is sometimes characterized as "HIV-negative AIDS", though, in fact, its clinical presentation differs somewhat from that seen with HIV/AIDS. [3] People with ICL have a weakened immune system and are susceptible to opportunistic infections , although the rate of infections is lower than in people with AIDS . [4] Contents 1 Cause 2 Pathophysiology 3 Diagnosis 4 Treatment 5 Prognosis 6 Epidemiology 7 References 8 External links Cause [ edit ] The cause of ICL, like all idiopathic conditions, is unknown. ... It has been associated with several cases of autoimmune disease Sjögren syndrome . [4] [20] Because all of the reported autoimmune diseases and lymphomas involve B cells , one hypothesis proposes that ICL's narrow T cell repertoire predisposes the immune system to B cell disorders. [4] Epidemiology [ edit ] ICL is a very rare disease . [2] In 1993, a total of 47 confirmed cases were reported in a survey sponsored by the Centers for Disease Control . [21] References [ edit ] ^ "OMIM Entry - # 615518 - IMMUNODEFICIENCY 13; IMD13" . omim.org . ... "Idiopathic CD4+ lymphocytopenia and Sjogren syndrome" (PDF) . Arch. Ophthalmol . 123 (7): 1012. doi : 10.1001/archopht.123.7.1012-a . ... "CD4+ T-lymphocytopenia--a frequent finding in anti-SSA antibody seropositive patients with primary Sjögren's syndrome" . J. Rheumatol . 31 (4): 726–8. ... External links [ edit ] Classification D ICD - 10 : D72.8 OMIM : 615518 MeSH : D018344 External resources Orphanet : 228000 v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency
Geschwind syndrome , also known as Gastaut-Geschwind , is a group of behavioral phenomena evident in some people with temporal lobe epilepsy . ... S2CID 22179745 . ^ a b Benson, D. F. (1991). "The Geschwind syndrome". Advances in Neurology . 55 : 411–21. ... "Psychopathological profile in patients with severe bilateral hippocampal atrophy and temporal lobe epilepsy: Evidence in support of the Geschwind syndrome?". Epilepsy & Behavior . 4 (3): 291–297. doi : 10.1016/S1525-5050(03)00084-2 . ... PMID 5480697 . ^ Waxman, Stephen G, MD; Geschwind, Norman, MD (1972). "The Interictal Behavior Syndrome of Temporal Lobe Epilepsy". Archives of General Psychiatry . 32 (12): 1580–1586. doi : 10.1001/archpsyc.1975.01760300118011 . ... Lay summary – specific symptoms that characterize the Geschwind syndrome like hypergraphia and hyposexuality might be pathogenically related to hippocampal atrophy . ^ Rees, Peter M; Fowler, Clare J; Maas, Cornelis (2007).