A progressive muscular dystrophy characterized by co-existence of limb-girdle weakness and diffuse joint contractures without cardiomyopathy. Patients present lower limb weakness progressing to involve also upper limbs and axial muscles and eventually leading to permanent loss of ambulation, widespread joint contractures in the limbs and sometimes the spine, and variable respiratory involvement. Morphological changes in muscle biopsies include rimmed vacuoles, increased internal nuclei, cytoplasmic bodies, and a dystrophic pattern.
A rare genetic neurodegenerative disease characterized by early-onset diffuse brain atrophy, growth failure with postnatal microcephaly, developmental delay, regression, profound intellectual disability, hypotonia, muscle weakness and atrophy, intractable seizures, spasticity, and optic atrophy. Patients are usually immobile and often require mechanical ventilation. Brain imaging shows cerebral and cerebellar atrophy, hypomyelination, and thin corpus callosum.
A number sign (#) is used with this entry because of evidence that early-onset progressive encephalopathy with brain atrophy and thin corpus callosum (PEBAT) is caused by homozygous or compound heterozygous mutation in the TBCD gene (604649) on chromosome 17q25. Description PEBAT is an autosomal recessive neurodevelopmental disorder characterized by severely delayed psychomotor development apparent soon after birth or in infancy, profound intellectual disability, poor or absent speech, and seizures. Most patients are never able to walk due to hypotonia or spasticity. Brain imaging shows cerebral and cerebellar atrophy, thin corpus callosum, and secondary hypomyelination. The disorder shows progressive features, including microcephaly, consistent with a neurodegenerative process (summary by Miyake et al., 2016; Flex et al., 2016). Clinical Features Miyake et al. (2016) reported 8 children from 4 unrelated families with a severe early-onset neurodegenerative encephalopathy.
A rare genetic skeletal muscle disease characterized by neonatal to childhood onset of slowly progressive muscle weakness and atrophy primarily affecting the lower limbs, joint contractures, kyphosis or lordosis of the spine, lateral tongue atrophy, and pes equinus. Progression to upper limb involvement, facial weakness, language impairment, intellectual disability, and behavioral abnormalities have been reported in addition. Muscle biopsy shows myopathic changes with increased fiber size variation, internalized nuclei, fiber atrophy, as well as rod structures and core targetoid defects.
A number sign (#) is used with this entry because of evidence that myofibrillar myopathy-7 (MFM7) is caused by homozygous mutation in the KY gene (605739) on chromosome 3q22. Description Myofibrillar myopathy-7 is an autosomal recessive muscle disorder characterized by early childhood onset of slowly progressive muscle weakness primary affecting the lower limbs and associated with joint contractures (summary by Straussberg et al., 2016). For a general phenotypic description and a discussion of genetic heterogeneity of myofibrillar myopathy, see MFM1 (601419). Clinical Features Straussberg et al. (2016) reported 2 adult brothers, born of consanguineous Israeli Arab parents, with onset of a slowly progressive myopathy from infancy. The lower limbs were primarily affected with weakness and muscle atrophy; upper limb involvement became apparent later, and both patients had atrophy of the lateral tongue margins.
A rare genetic neurological disorder characterized by early-onset severe global developmental delay with regression, congenital or acquired microcephaly, hearing loss, truncal hypotonia, appendicular spasticity, and dystonia and/or myoclonus. Additional reported manifestations include seizures, optic atrophy, cortical visual impairment, scoliosis, and dysphagia. Brain imaging shows pontine hypoplasia, partial agenesis of the corpus callosum, and diffuse cerebral atrophy with relative sparing of the cerebellum.
A number sign (#) is used with this entry because of evidence that early-onset progressive encephalopathy with brain atrophy and spasticity (PEBAS) is caused by homozygous or compound heterozygous mutation in the TRAPPC12 gene (614139) on chromosome 2p25. Clinical Features Milev et al. (2017) reported 3 children from 2 unrelated families with severe early-onset progressive encephalopathy characterized by microcephaly, global developmental delay, and hearing loss. One patient, born of consanguineous Palestinian parents, presented with brief flexion seizures at age 5 months and subsequently showed developmental regression and stagnation with loss of smiling, visual tracking, and social overtures. EEG showed hypsarrhythmia. Family history in this patient was notable for the mother having 5 previous pregnancies ending in spontaneous abortions, a sixth terminated for multiple anomalies, and another child born at 24 weeks' gestation who died soon after birth. In the second family, 2 sisters were similarly affected. Both had small developmental gains in infancy, but then plateaued or showed regression and had severe global developmental deficits.
Fallot complex - intellectual deficit - growth delay is a rare disorder characterized by tetralogy of Fallot, minor facial anomalies, and severe intellectual deficiency and growth delay. Epidemiology To date, five patients have been reported in two families. Clinical description Dysmorphic features include large, protruding, abnormally modeled ears and broad nasal root. Microcephaly and syndactyly of 2nd and 3rd toes have also been recorded. All five patients have severe intellectual deficiency. Genetic counseling The condition is probably hereditary, and is transmitted as an autosomal recessive trait.
Bindewald et al. (1994) described an apparently novel autosomal recessive syndrome in 3 brothers and a sister in a Pakistani family with first-cousin parents. ... Tetralogy of Fallot has been observed in association with a number of syndromes and as an isolated familial occurrence.
A rare systemic disease characterized by generalized joint hypermobility with recurrent joint dislocations, redundant and hyperextensible skin with poor wound healing and abnormal scarring, easy bruising, and osteopenia/osteoporosis. Additional manifestations include hypotonia, delayed motor development, foot deformities, prominent superficial veins in the chest region, vascular complications (like mitral valve prolapse and aortic root dilation), hernias, dental anomalies, scoliosis, and facial dysmorphisms (like high palate, micrognathia, narrow palate). Mode of inheritance is autosomal recessive.
A number sign (#) is used with this entry because of evidence that Ehlers-Danlos syndrome classic-like-2 (EDSCLL2) is caused by homozygous or compound heterozygous mutation in the AEBP1 gene (602981) on chromosome 7p13. Description Ehlers-Danlos syndrome classic-like-2 is characterized by severe joint and skin laxity, osteoporosis involving the hips and spine, osteoarthritis, soft redundant skin that can be acrogeria-like, delayed wound healing with abnormal atrophic scarring, and shoulder, hip, knee, and ankle dislocations. ... See 606408 for another classic-like EDS syndrome. For a discussion of the classification of EDS, see 130000. ... In 2 unrelated men with classic-like Ehlers-Danlos syndrome, Blackburn et al. (2018) identified compound heterozygosity and homozygosity, respectively, for mutations in the AEBP1 gene (602981.0002-602981.0004).
A rare genetic neuromuscular disease characterized by early onset of proximal or generalized muscle weakness, external ophthalmoplegia with or without ptosis, and joint contractures. Hypotonia, neonatal respiratory distress necessitating ventilation, and severe dysphagia have also been reported. The disease is of variable severity and non- or slowly progressive. Patients typically remain ambulatory. Muscle biopsy may show predominance of type 1 fibers, marked variability in fiber size, increased internal nuclei, and proliferation of perimysial and endomysial connective tissue.
A number sign (#) is used with this entry because of evidence that proximal myopathy and ophthalmoplegia (MYPOP) is caused by heterozygous, compound heterozygous, or homozygous mutation in the gene encoding myosin heavy chain IIa (MYHC2A, or MYH2; 160740) on chromosome 17p13. Description Proximal myopathy and ophthalmoplegia is a relatively mild muscle disorder characterized by childhood onset of symptoms. The disorder is either slowly progressive or nonprogressive, and affected individuals retain ambulation, although there is variable severity. MYPOP can show both autosomal dominant and autosomal recessive inheritance; the phenotype is similar in both forms (summary by Lossos et al., 2005 and Tajsharghi et al., 2014). Clinical Features Autosomal Dominant Inheritance Darin et al. (1998) described a multigeneration Swedish family with an apparently novel myopathy inherited as an autosomal dominant.
A rare, genetic, non-dystrophic myopathy disease characterized by childhood-onset severe external ophthalmoplegia, typically without ptosis, associated with mild, very slowly progressive muscular weakness and atrophy, involving the facial, neck flexor and limb (upper > lower, proximal > distal) muscles. Muscle biopsy shows type 1 fiber uniformity, absent, or abnormally small, type 2A fibers, increased variability of fiber size, internalized nuclei and/or fatty infiltration.
A rare, severe, genetic, neurometabolic disease characterized by infantile-onset of progressive neurodevelopmental regression, optic atrophy with nystagmus and diffuse white matter disease. Affected individuals usually have central hypotonia that progresses to limb spasticity and hyperreflexia, eventually resulting in a vegetative state. Recurrent chest infections are frequently associated and seizures (usually generalized tonic-clonic) may occasionally be observed. Brain magnetic resonance imaging shows diffuse bilateral symmetric abnormalities in the cerebral periventricular white matter, with variable lesions in other areas but sparing the basal ganglia.
A number sign (#) is used with this entry because of evidence that multiple mitochondrial dysfunctions syndrome-4 (MMDS4) is caused by homozygous or compound heterozygous mutation in the ISCA2 gene (615317) on chromosome 14q24. ... For a general description and a discussion of genetic heterogeneity of multiple mitochondrial dysfunctions syndrome, see MMDS1 (605711). Clinical Features Al-Hassnan et al. (2015) reported 5 unrelated consanguineous Arab families in which 6 children had a severe neurologic disorder characterized by onset of neuroregression between ages 3 and 7 months.
A rare genetic neurodegenerative disease characterized by neonatal to infantile onset of hypotonia, developmental delay, regression of motor skills with distal amyotrophy, ataxia, and spasticity, absent speech or dysarthria, and moderate to severe cognitive impairment. Optic atrophy may also be associated. Brain imaging shows cerebellar atrophy and thin corpus callosum, as well as brain iron accumulation in the pallidum and substantia nigra beginning during the second decade of life.
Biallelic mutation in the TBCE gene can also cause hypoparathyroidism-retardation-dysmorphism syndrome (HRDS; 241410) and Kenny-Caffey syndrome (KCS1; 244460).
A rare mitochondrial disease characterized by prenatal complications including oligohydramnios, fetal growth restriction, hydrops, and anemia, followed by severe lactic acidosis, hyaline membrane disease, pulmonary hypertension, cardiac anomalies, liver dysfunction, urogenital abnormalities and progressive renal disease, seizures, thrombocytopenia, and sideroblastic anemia resulting in multisystem organ failure and death shortly after birth. Less severely affected patients surviving the neonatal period and showing sensorineural hearing loss and developmental delay have been reported.
A number sign (#) is used with this entry because of evidence that hydrops, lactic acidosis, and sideroblastic anemia (HLASA) is caused by compound heterozygous mutation in the LARS2 gene (604544) on chromosome 3p21. One such patient has been reported. Clinical Features Riley et al. (2016) reported a female infant, born of unrelated Pakistani parents, with a lethal multisystem disorder resulting in death at 5 days of age. The pregnancy was complicated by oligohydramnios, fetal growth restriction, hydrops, and anemia, with antenatal scans showing fetal pericardial effusion, ascites, and scalp edema. The infant was delivered prematurely at 29 weeks by emergency cesarean section, and intubated and ventilated from birth. There were multisystem complications, including severe metabolic acidosis with increased lactate, hyaline membrane disease, mild cardiac defects associated with tachyarrhythmias, pulmonary hypertension, thrombocytopenia, and anemia.
However, this kidney tends to be hypertrophied, ectopic and prone to infection and damage. [4] It may be associated with an increased incidence of Müllerian duct abnormalities, which are abnormalities of the development of the female reproductive tract and can be a cause of infertility , blocked menstrual flow ( hematocolpos ), increased need for Caesarean sections , or other problems. Herlyn-Werner-Wunderlich syndrome is one such syndrome in which unilaterial renal agenesis is combined with a blind hemivagina and uterus didelphys . [5] Up to 40% of women with a urogenital tract anomaly also have an associated renal tract anomaly. [6] Adults with unilateral renal agenesis have considerably higher chances of hypertension (high blood pressure). ... A possible complication later in life of unilateral renal agenesis is Focal Segmental Glomerular Sclerosis (FSGS) which will cause nephrotic syndrome, potentially resulting from glomerular overload. [8] Genetics [ edit ] In 2008 researchers found autosomal dominant mutations in the RET and GDNF genes to be linked to renal agenesis in unrelated stillborn fetuses through PCR and direct sequence analysis. [9] In the study, DNA from 33 stillborn fetuses were sequenced for mutations in RET, GDNF and GFRA1 . ... However, renal agenesis and other causes of oligohydramnios sequence have been linked to a number of other conditions and syndromes to include Down syndrome , Kallmann syndrome , branchio-oto-renal syndrome and others. ... Springer. p. 22. ^ Ahmad, Zohra; Goyal, Ankur; Das, Chandan J; Deka, Dipika; Sharma, Raju (2013-01-01). "Herlyn–Werner–Wunderlich syndrome presenting with infertility: Role of MRI in diagnosis" . ... External links [ edit ] Classification D ICD - 10 : Q60.0 - Q60.2 ICD - 9-CM : 753.0 OMIM : 191830 DiseasesDB : 11252 v t e Congenital malformations and deformations of urinary system Abdominal Kidney Renal agenesis / Potter sequence , Papillorenal syndrome cystic Polycystic kidney disease Meckel syndrome Multicystic dysplastic kidney Medullary sponge kidney Horseshoe kidney Renal ectopia Nephronophthisis Supernumerary kidney Pelvic kidney Dent's disease Alport syndrome Ureter Ectopic ureter Megaureter Duplicated ureter Pelvic Bladder Bladder exstrophy Urethra Epispadias Hypospadias Posterior urethral valves Penoscrotal transposition Vestigial Urachus Urachal cyst Urachal fistula Urachal sinus
Generally, it may occur as an isolated condition or as a component of numerous rare syndromes, either secondary due to antenatal urine obstruction or due to primary maldevelopment of nephrogenic tissues. ... Incomplete penetrance and variable expressivity are very common in CAKUT (congenital anomalies of kidney and urinary tract); thus, genes involved in multi-organ syndromes ( such as EYA1 , GATA3 , GREBI1L , PAX2 , PBX1 , SALL1 , FRAS1 , FREM2 , and GRIP1 ) may possibly lead to an isolated renal phenotype (dysplasia/hypoplasia/agenesis). ... Renal dysplasia may occur as part of recognized syndromes such as Kallmann, Bardet-Biedl, Beckwith-Wiedemann, diGeorge, Fraser, renal coloboma, and renal cysts and diabetes syndromes. ... Genetic counseling Families may benefit from genetic counseling in case of renal dysplasia as part of a multi-organ syndrome, in familial cases or in bilateral cases with chronic kidney disease.
They are associated with increased vulnerability to infection, but can be difficult to detect (or asymptomatic) in the absence of infection. [ citation needed ] Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 4 Treatment 5 See also 6 References 7 Further reading 8 External links Signs and symptoms [ edit ] Signs/symptoms of humoral immune deficiency depend on the cause, but generally include signs of infection such as: [1] Sinusitis Sepsis Skin infection Pneumonia Causes [ edit ] Cause of this deficiency is divided into primary and secondary : Primary the International Union of Immunological Societies classifies primary immune deficiencies of the humoral system as follows: [3] [2] Hyper-IgM syndromes(immunoglobulin M) Absent B cells with a resultant severe reduction of all types of antibody: X-linked agammaglobulinemia ( btk deficiency, or Bruton 's agammaglobulinemia), μ - Heavy chain deficiency, l 5 deficiency, Igα deficiency, BLNK deficiency, thymoma with immunodeficiency B cells low but present, but with reduction in 2 or more isotypes (usually IgG & IgA, sometimes IgM): common variable immunodeficiency (CVID), ICOS deficiency, CD19 deficiency, TACI (TNFRSF13B) deficiency, BAFF receptor deficiency. Normal numbers of B cells with decreased IgG and IgA and increased IgM : Hyper-IgM syndromes Normal numbers of B cells with isotype or light chain deficiencies: heavy chain deletions, kappa chain deficiency, isolated IgG subclass deficiency, IgA with IgG subsclass deficiency, selective immunoglobulin A deficiency Transient hypogammaglobulinemia of infancy (THI) Secondary secondary (or acquired) forms of humoral immune deficiency are mainly due to hematopoietic malignancies and infections that disrupt the immune system: [4] Multiple myeloma Chronic lymphoid leukemia AIDS Diagnosis [ edit ] Human B cell In terms of diagnosis of humoral immune deficiency depends upon the following: [5] [6] Measure serum immunoglobulin levels B cell count Family medical history Treatment [ edit ] Further information: Immunoglobulin therapy Treatment for B cell deficiency (humoral immune deficiency) depends on the cause, however generally the following applies: [5] Treatment of infection (antibiotics) Surveillance for malignancies Immunoglobulin replacement therapy See also [ edit ] Immunodeficiency T cell deficiency References [ edit ] ^ a b N. ... External links [ edit ] PubMed Classification D ICD - 10 : D80 ICD - 9-CM : 279.0 Scholia has a topic profile for Humoral immune deficiency . v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
It is difficult to distinguish from, and has been found to co-occur with, alexithymia . [2] Contents 1 Presentation 2 Causes 3 Diagnosis 3.1 Classification 3.1.1 Faux Pas test 3.1.2 Strange Stories test 3.1.3 Other tests 3.2 Differential diagnosis 3.2.1 Autism or Asperger's syndrome 3.2.2 Klüver–Bucy syndrome 4 Research 5 References Presentation [ edit ] Damage to the right temporal occipital region in humans has been associated with the inability to recognize the faces of loved ones, friends, and pets (considered a form of prosopagnosia ). [3] This limits the ability to appropriately interact with familiar people, potentially severely damaging interpersonal relationships. ... Other tests [ edit ] Another test that could be used to diagnose emotional deficits is the Facial Recognition Test, where subjects are presented a number of pictures of faces with a variety of expressions, and are asked to determine what emotion they are depicting. [11] Differential diagnosis [ edit ] The constellation of symptoms in social-emotional agnosia can also be seen in a number of different behavioral disorders. Autism or Asperger's syndrome [ edit ] Both autism and Asperger's syndrome show deficits in understanding others' mental states, including the recognition of emotional expressions. Damage to the amygdala has also been implicated for these disorders, which can explain why the symptoms appear to overlap. [4] Klüver–Bucy syndrome [ edit ] Although rare in humans, Klüver–Bucy syndrome has many symptoms that are strikingly similar to those seen in social-emotional agnosia. The amygdala and temporal lobes have been implicated in the pathology of Klüver–Bucy syndrome as well, leading to docility, hyperorality, and in some rare cases hypersexuality. ... Monkeys with Klüver–Bucy syndrome demonstrated a loss of fear and aggression, hyperorality, and hypersexuality.
Involvement of T1 may result in Horner's syndrome , with ptosis , and miosis . Weakness or lack of ability to use specific muscles of the shoulder or arm. [1] [11] [12] It can be contrasted to Erb-Duchenne's palsy , which affects C5 and C6 . ... If Horner syndrome is present, there is miosis (constriction of the pupils) in the affected eye. [ citation needed ] The injury can result from difficulties in childbirth . ... Lower brachial plexus injuries should be distinguished from upper brachial plexus injuries, which can also result from birth trauma but give a different syndrome of weakness known as Erb's palsy . ... "Histopathological basis of Horner's syndrome in obstetric brachial plexus palsy differs from that in adult brachial plexus injury". ... ISBN 978-0-8016-3227-3 . pp.576, 667 ^ a b Page 512: Lower Radicular Syndrome (Klumpke Paralysis) in: Pedley, Timothy A.; Rowland, Lewis P.; Merritt, Hiram Houston (2010).
This symptom may also be referred to as Horner syndrome . Klumpke paralysis is caused by an injury to the nerves of the brachial plexus that which may result during birth due to a a difficult delivery.
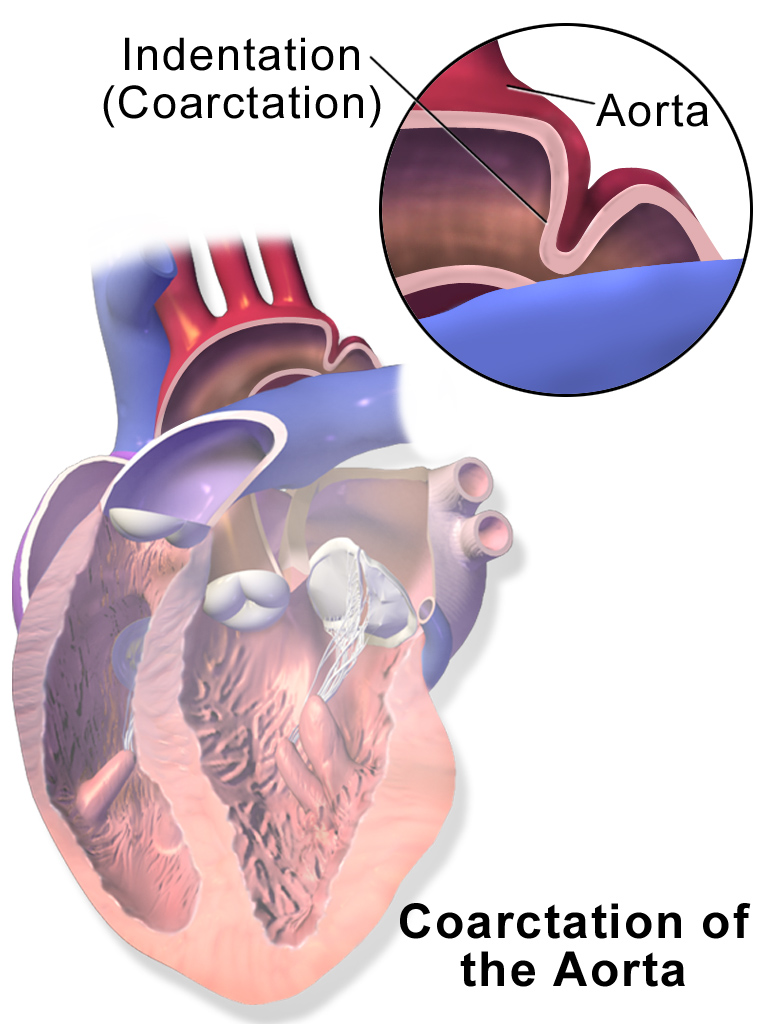
The heart defects classified as CCHD include coarctation of the aorta , double-outlet right ventricle, D-transposition of the great arteries , Ebstein anomaly, hypoplastic left heart syndrome , interrupted aortic arch, pulmonary atresia with intact septum , single ventricle, total anomalous pulmonary venous connection , tetralogy of Fallot , tricuspid atresia , and truncus arteriosus . ... However, the heart defects associated with CCHD can also occur as part of genetic syndromes that have additional features. Some of these genetic conditions, such as Down syndrome, Turner syndrome, and 22q11.2 deletion syndrome, result from changes in the number or structure of particular chromosomes. Other conditions, including Noonan syndrome and Alagille syndrome, result from mutations in single genes.
Having certain genetic conditions, such as Turner syndrome, also raises the risk of coarctation of the aorta. ... Talk to your health care provider if you or your child has a condition that increases the risk of aortic coarctation, such as Turner syndrome, bicuspid aortic valve or another heart defect.
This is the type seen in approximately 5% of infants with Turner syndrome . [4] [5] Ductal coarctation: The narrowing occurs at the insertion of the ductus arteriosus. ... It is common in girls who have Turner syndrome . Symptoms may be absent with mild narrowings (coarctation). ... Some signs that can lead to a coarctation have been linked to pathologies such as Turner syndrome , bicuspid aortic valve, and other family heart conditions. [5] Treatment [ edit ] In adults and children found to have coarctation, treatment is conservative if asymptomatic, but may require surgical resection of the narrow segment if there is arterial hypertension . ... "Cardiovascular anomalies in children and young adults with Ullrich-Turner syndrome-the erlangen experience" . Clinical Cardiology . 28 (2): 88–92. doi : 10.1002/clc.4960280209 . ... Classification D ICD - 10 : Q25.1 ICD - 9-CM : 747.10 OMIM : 120000 MeSH : D001017 DiseasesDB : 2876 External resources MedlinePlus : 000191 eMedicine : med/154 Patient UK : Coarctation of the aorta v t e Congenital vascular defects / Vascular malformation Great arteries / other arteries Aorta Patent ductus arteriosus Coarctation of the aorta Interrupted aortic arch Double aortic arch Right-sided aortic arch Overriding aorta Aneurysm of sinus of Valsalva Vascular ring Pulmonary artery Pulmonary atresia Stenosis of pulmonary artery Subclavian artery Aberrant subclavian artery Umbilical artery Single umbilical artery Great veins Superior / inferior vena cava Congenital stenosis of vena cava Persistent left superior vena cava Pulmonary vein Anomalous pulmonary venous connection ( Total , Partial ) Scimitar syndrome Arteriovenous malformation Cerebral arteriovenous malformation
Gough (1961) described the anomaly in father and son. He found 6 other reports of familial coarctation. In a systematic study of coarctation, Boon and Roberts (1976) discerned familial aggregation with multifactorial inheritance. Recurrence risks in sibs was about 0.5% for coarctation and 1.0% for any form of congenital heart defect. Beekman and Robinow (1985) described coarctation of the aorta in 4 generations. In 2 members of the family, mother and son, in the third and fourth generations, the coarctation was minimal; in the mother, for example, a gradient in the aorta was demonstrated mainly after peak exercise.
A number sign (#) is used with this entry because of evidence that Hermansky-Pudlak syndrome-10 (HPS10) is caused by homozygous mutation in the AP3D1 gene (607246) on chromosome 19p13. One such patient has been reported. Description Hermansky-Pudlak syndrome-10 is an autosomal recessive multisystem disorder characterized by infantile onset of immunodeficiency, oculocutaneous albinism, and severe neurologic impairment, including severely delayed global development and intractable seizures (summary by Ammann et al., 2016). For a general phenotypic description and a discussion of genetic heterogeneity of Hermansky-Pudlak syndrome, see HPS1 (203300). Clinical Features Ammann et al. (2016) reported a boy, born of consanguineous Turkish parents, with severe neurologic impairment, albinism, and immunodeficiency. ... Animal Model The 'mocha' mouse mutant, a model for Hermansky-Pudlak syndrome (203300), shows coat and eye color dilution, reduced levels of renal lysosomal enzymes in urine, and prolonged bleeding due to storage pool deficiency in the dense granules of platelets.
Congenital hypothyroidism can also occur as part of syndromes that affect other organs and tissues in the body. These forms of the condition are described as syndromic. Some common forms of syndromic hypothyroidism include Pendred syndrome, Bamforth-Lazarus syndrome, and brain-lung-thyroid syndrome. ... Still other genes are involved in syndromic forms of the disorder. Learn more about the genes associated with Congenital hypothyroidism DUOX2 PAX8 SLC26A4 SLC5A5 TG TPO TSHB TSHR Additional Information from NCBI Gene: DUOXA2 IYD NKX2-5 THRA TRHR Inheritance Pattern Most cases of congenital hypothyroidism are sporadic, which means they occur in people with no history of the disorder in their family.
A type of primary congenital hypothyroidism, a permanent thyroid hormone deficiency that is present from birth due to thyroid resistance to TSH. Epidemiology Resistance to TSH occurs in about 5% of cases of permanent congenital hypothyroidism. Clinical description Clinical manifestations are those of other forms of congenital hypothyroidism (CH; see this term). Goiter is always absent. Etiology Mutations in the TSH receptor gene ( TSHR ; 14q31) result in resistance to TSH, which causes a reduction in thyroid hormone production. Mutations in TSHR may also cause thyroid hypoplasia (see this term).
A number sign (#) is used with this entry because of evidence that congenital nongoitrous hypothyroidism-1 (CHNG1) is caused by homozygous or compound heterozygous mutation in the gene encoding the thyroid-stimulating hormone receptor (TSHR; 603372) on chromosome 14q31. Description Resistance to thyroid-stimulating hormone (TSH; see 188540), a hallmark of congenital nongoitrous hypothyroidism, causes increased levels of plasma TSH and low levels of thyroid hormone. Only a subset of patients develop frank hypothyroidism; the remainder are euthyroid and asymptomatic (so-called compensated hypothyroidism) and are usually detected by neonatal screening programs (Paschke and Ludgate, 1997). Genetic Heterogeneity of Congenital Nongoitrous Hypothyroidism Also see CHNG2 (218700), caused by mutation in the PAX8 gene (167415) on chromosome 2q14; CHNG3 (609893), mapped to chromosome 15q25.3; CHNG4 (275100), caused by mutation in the TSHB gene (188540) on chromosome 1p13; CHNG5 (225250), caused by mutation in the NKX2-5 gene (600584) on chromosome 5q35; CHNG6 (614450), caused by mutation in the THRA gene (190120) on chromosome 17q21; and CHNG7 (618573), caused by mutation in the TRHR gene (188545) on chromosome 8q24. Clinical Features Stanbury et al. (1968) described an 8-year-old boy with congenital hypothyroidism who was the offspring of parents related as first cousins once removed.