In 12 (27%) of the 44 patients, Ebstein anomaly was part of a syndrome, and 7 of those patients were diagnosed with distinct disorders, including CHARGE syndrome (214800) in 2, and VACTERL association (see 192350), Noonan syndrome (163950), Kabuki syndrome (147920), Holt-Oram syndrome (142900), and Cornelia de Lange syndrome (see 122470) in 1 each. ... In a study of 44 consecutive patients with Ebstein anomaly, Digilio et al. (2011) performed standard chromosome analysis and array CGH in the 12 patients in whom the anomaly was part of a syndrome, and identified chromosomal anomalies in 3 of them: a 1p36 deletion (see 607872) in association with an Xpter-Xp22.3 duplication (see 300830), an 8p23.1 deletion, and a deletion of 18q21.3-qter (see 601808).
Changes in the heartbeat can make it harder for the heart to work as it should. Wolff-Parkinson-White (WPW) syndrome. In this condition, an extra signaling pathway between the heart's upper and lower chambers causes a fast heartbeat and fainting.
During adulthood, supraventricular tachycardia can also be observed, a proportion of patients also having Wolff-Parkinson-White syndrome (see this term). The malformation is often associated with other cardiac lesions, such as atrial or ventricular septal defects, patency of the arterial duct, and pulmonary stenosis.
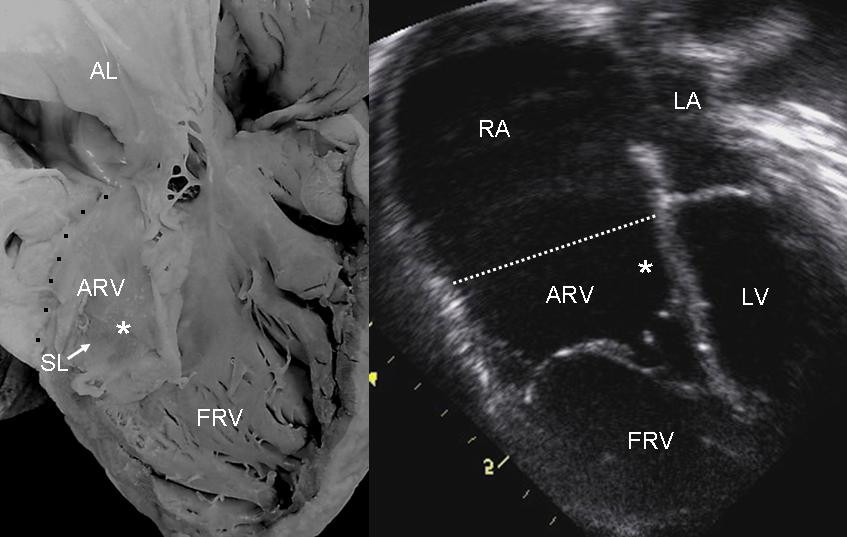
S3 heart sound S4 heart sound Triple or quadruple gallop due to widely split S1 and S2 sounds plus a loud S3 and/or S4 Systolic murmur of tricuspid regurgitation = Holosystolic or early systolic murmur along the lower left sternal border depending on the severity of the regurgitation Right atrial hypertrophy Right ventricular conduction defects Wolff-Parkinson-White syndrome often accompanies Related abnormalities [ edit ] A diagram showing the downward displacement of the tricuspid valve from its normal position in the fibrous ring down into the right ventricle. ... About 50% of individuals with Ebstein's anomaly have an associated shunt between the right and left atria, either an atrial septal defect or a patent foramen ovale . [3] Electrophysiologic abnormalities [ edit ] About 50% of individuals with Ebstein's anomaly have an accessory pathway with evidence of Wolff-Parkinson-White syndrome, secondary to the atrialized right ventricular tissue. ... Other abnormalities that can be seen on the ECG include: signs of right atrial enlargement or tall and broad 'Himalayan' P waves first degree atrioventricular block manifesting as a prolonged PR-interval low amplitude QRS complexes in the right precordial leads atypical right bundle branch block T wave inversion in V1-V4 and Q waves in V1-V4 and II, III and aVF. [4] Risk factors [ edit ] An enlargement of the aorta may occur; an increased risk of abnormality is seen in babies of women taking lithium during the first trimester of pregnancy [5] (though some have questioned this) [6] and in those with Wolff-Parkinson-White syndrome . Treatment [ edit ] Medication [ edit ] Ebstein's cardiophysiology typically presents as an (antidromic) AV reentrant tachycardia with associated pre-excitation . ... External links [ edit ] Classification D ICD - 10 : Q22.5 ICD - 9-CM : 746.2 OMIM : 224700 MeSH : D004437 DiseasesDB : 4039 External resources MedlinePlus : 007321 eMedicine : med/627 Wikimedia Commons has media related to Ebstein's anomaly . v t e Congenital heart defects Heart septal defect Aortopulmonary septal defect Double outlet right ventricle Taussig–Bing syndrome Transposition of the great vessels dextro levo Persistent truncus arteriosus Aortopulmonary window Atrial septal defect Sinus venosus atrial septal defect Lutembacher's syndrome Ventricular septal defect Tetralogy of Fallot Atrioventricular septal defect Ostium primum Consequences Cardiac shunt Cyanotic heart disease Eisenmenger syndrome Valvular heart disease Right pulmonary valves stenosis insufficiency absence tricuspid valves stenosis atresia Ebstein's anomaly Left aortic valves stenosis insufficiency bicuspid mitral valves stenosis regurgitation Other Underdeveloped heart chambers right left Uhl anomaly Dextrocardia Levocardia Cor triatriatum Crisscross heart Brugada syndrome Coronary artery anomaly Anomalous aortic origin of a coronary artery Ventricular inversion
Ebstein's anomaly is a rare heart defect in which parts of the tricuspid valve (which separates the right ventricle from the right atrium) are abnormal. The abnormality causes the tricuspid valve to leak blood backwards into the right atrium. The backup of blood flow can lead to heart swelling and fluid buildup in the lungs or liver. Sometimes, not enough blood gets out of the heart into the lungs and the person may appear blue. Symptoms range from mild to very severe. Treatment depends on the severity of the defect and may include medications, oxygen therapy, or surgery.
Chisa (1965) reported affected brother and sister, and Kopf et al. (1965) described affected sisters. Herd and Hunter (1998) quoted an incidence of halo nevi of 1%. They reported a family that had lived in New Zealand in which a typical halo nevus was noted below the left breast of the mother. One daughter, aged 9 years, had 2 halo nevi, and 2 older daughters had 1 or 2 halo nevi. All 4 individuals had had much sun exposure.
A number sign (#) is used with this entry because of evidence that pseudo-TORCH syndrome-2 (PTORCH2) is caused by homozygous or compound heterozygous mutation in the USP18 gene (607057) on chromosome 22q11. Description Pseudo-TORCH syndrome-2 is an autosomal recessive multisystem disorder characterized by antenatal onset of intracranial hemorrhage, calcification, brain malformations, liver dysfunction, and often thrombocytopenia. ... Clinical Features Knoblauch et al. (2003) described 2 brothers, born to healthy nonconsanguineous parents of German origin, who showed features resembling congenital intrauterine infection-like syndrome. Both showed extensive intra- and extracranial calcifications, thrombocytopenia, a septum pellucidum cyst, 1-sided paresis of the diaphragm, metaphyseal changes on x-ray scans resembling those produced by intrauterine infection, and hepatosplenomegaly. ... The authors thought that this disorder could be distinguished from Aicardi-Goutieres syndrome (see, e.g., AGS1, 225750). Meuwissen et al. (2016) reported follow-up of the patients reported by Knoblauch et al. (2003), noting that both sibs had thrombocytopenia and that one had dyserythropoiesis on bone marrow aspiration.
A rare genetic neurological disorder characterized by severe pseudo-TORCH syndrome with signs of brain damage and occasionally systemic manifestations resembling the sequelae of congenital infection, but in the absence of an infectious agent.
Factor V Leiden thrombophilia is an inherited disorder of blood clotting . Factor V Leiden is the name of a specific gene mutation that results in thrombophilia, which is an increased tendency to form abnormal blood clots that can block blood vessels. People with factor V Leiden thrombophilia have a higher than average risk of developing a type of blood clot called a deep venous thrombosis (DVT). DVTs occur most often in the legs, although they can also occur in other parts of the body, including the brain, eyes, liver, and kidneys. Factor V Leiden thrombophilia also increases the risk that clots will break away from their original site and travel through the bloodstream.
The clinical expression of factor V Leiden thrombophilia is influenced by the following: The number of Leiden variants (heterozygotes have a slightly increased risk for venous thrombosis; homozygotes have a much greater thrombotic risk) Coexisting genetic thrombophilic disorders, which have a supra-additive effect on overall thrombotic risk Acquired thrombophilic disorders: antiphospholipid antibody (APLA) syndrome, paroxysmal nocturnal hemoglobinuria, myeloproliferative disorders, and increased levels of clotting factors Circumstantial risk factors including but not limited to pregnancy, central venous catheters, travel, combined oral contraceptive (COC) use and other combined contraceptives, oral hormone replacement therapy (HRT), selective estrogen receptor modulators (SERMs), obesity, leg injury, and advancing age Diagnosis/testing. ... A heterozygous Leiden variant was associated with the following: A sixfold increased risk for primary upper-extremity thrombosis (not related to malignancy or a venous catheter) [Martinelli et al 2004] A sixfold increased risk of superficial vein thrombosis not associated with varicose veins, malignancy, or autoimmune disorders [Martinelli et al 1999] Increased risk of venous thrombosis at unusual sites [Martinelli et al 2014] A fourfold increased risk of cerebral venous thrombosis [Dentali et al 2006] A Leiden variant: May increase the risk of splanchic vein thrombosis; Was associated with an 11-fold increased risk of Budd-Chiari syndrome in case-control studies [Janssen et al 2000]; Confers a threefold increased risk of portal vein thrombosis (meta-analysis by Dentali et al [2008]). ... Acquired thrombophilic disorders include antiphospholipid antibody (APLA) syndrome, paroxysmal nocturnal hemoglobinuria, myeloproliferative disorders, and increased levels of clotting factors. ... Management Evaluations Following Initial Diagnosis To assess the risk for thrombosis in an individual found to have the factor V Leiden variant, the following evaluations are recommended: For individuals heterozygous for the Leiden variant: the following testing for other inherited or acquired thrombophilic disorders is recommended by experts (but is not a hard-and-fast rule) given that double heterozygosity for the Leiden variant and F2 thrombophilia variant 20210G>A occurs more commonly than protein C, S, and AT deficiencies (which are rare and unlikely to be found except in those with "high risk features" such as a strong family history) and antiphospholipid antibody (APLA) syndrome can occur at any age in anyone: DNA testing F2 thrombophilia variant (c.*97G>A, commonly known as 20210G>A) Multiple phospholipid-dependent coagulation assays for a lupus inhibitor Serologic assays for anticardiolipin antibodies and anti-beta 2 -glycoprotein 1 antibodies For high-risk individuals (i.e., those with a history of recurrent VTE, especially at young age, or those with strong family history of VTE at young age) evaluation should also include assays of: Protein C activity Antithrombin activity Protein S activity or free protein S antigen Note: Measurement of the following is NOT recommended: Plasma concentration of homocysteine since no data support a change in duration of anticoagulation or the use of vitamin supplementation in individuals with hyperhomocysteinemia and a history of VTE MTHFR variants as no clinical rationale for this testing exists Factor VIII and other clotting factor levels [Moll 2015] Treatment of Manifestations Treatment of VTE in Adults The management of individuals with factor V Leiden thrombophilia depends on the clinical circumstances.
The HELLP syndrome is a severe presentation of preeclampsia (see 189800). The finding of the R506Q mutation suggested that the pathogenesis of HELLP syndrome may be associated with a thrombotic process. ... Gurakan et al. (1999) described a child with Budd-Chiari syndrome who was homozygous for the factor V Leiden mutation. ... As many as 50% of patients with Budd-Chiari syndrome have a myeloproliferative disorder, either preexisting or diagnosed at the time of the syndrome. However, some patients with the Budd-Chiari syndrome may have a latent myeloproliferative disorder without elevated blood counts.
Researchers have identified several forms of CFEOM, designated CFEOM1, CFEOM2, CFEOM3, and Tukel syndrome (sometimes called CFEOM4). The specific problems with eye movement vary among the types, and some types are associated with additional signs and symptoms. ... CFEOM3 can include additional neurological problems, such as intellectual disability; difficulty with social skills; a smaller-than-normal head size (microcephaly ); muscle weakness in the face; nonfunctioning vocal cords; and a set of symptoms called Kallmann syndrome, which features delayed or absent puberty and an impaired sense of smell. ... CFEOM1 and CFEOM3 have been reported worldwide, whereas CFEOM2 has been seen in only a few families of Turkish, Saudi Arabian, and Iranian descent. Tukel syndrome appears to be very rare; it has been diagnosed in only one large Turkish family. ... Studies suggest that a gene associated with Tukel syndrome may be located near one end of chromosome 21. ... The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Tukel syndrome also appears to have an autosomal recessive pattern of inheritance, although the genetic change responsible for this disorder is unknown.
Cytokine storm syndromes include familiar hemophagocytic lymphohistiocytosis , Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis, systemic or non-systemic juvenile idiopathic arthritis –associated macrophage activation syndrome , NLRC4 macrophage activation syndrome, cytokine release syndrome and sepsis . [6] Contents 1 Cytokine storms versus cytokine release syndrome 2 Research 3 History 3.1 Relationship to COVID-19 4 References Cytokine storms versus cytokine release syndrome [ edit ] The term "cytokine storm" is often loosely used interchangeably with cytokine release syndrome (CRS) but is more precisely a differentiable syndrome that may represent a severe episode of cytokine release syndrome or a component of another disease entity, such as macrophage activation syndrome . ... PMID 32531256 . ^ Behrens, Edward M.; Koretzky, Gary A. (2017). "Review: Cytokine Storm Syndrome: Looking Toward the Precision Medicine Era" . ... PMID 28217930 . ^ Porter D, Frey N, Wood PA, Weng Y, Grupp SA (March 2018). "Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel" . ... "High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome" . The Journal of Infectious Diseases . 179 (2): 295–302. doi : 10.1086/314597 . ... "COVID-19: consider cytokine storm syndromes and immunosuppression" . Lancet . 395 (10229): 1033–1034. doi : 10.1016/S0140-6736(20)30628-0 .
The authors referred to the disorder as the Matthew-Wood syndrome at the request of the parents, presumably the name of the first-born sib. ... Molecular Genetics In 5 families with syndromic clinical anophthalmia, Pasutto et al. (2007) detected homozygosity for mutations in the STRA6 gene (610745). ... STRA6 mutations thus define a pleiotropic malformation syndrome representing the first human phenotype associated with mutations in a gene from the STRA group. Pasutto et al. (2007) suggested that the clinical anophthalmia syndrome that they described and showed to be caused by mutations in the STRA6 gene may be the same disorder as that known as Matthew-Wood syndrome. ... Martinovic-Bouriel et al. (2007) studied 2 familial cases of Matthew-Wood syndrome and excluded mutations in the FGF10 (602115) and FGFR2 (see 176943) genes.
Their parents had normal eyes upon examination, and there were 2 other unaffected sisters. Syndromic Microphthalmia 15 Chassaing et al. (2016) reported a 9-year-old boy, born of first-cousin parents, who had bilateral colobomatous microphthalmia noted at birth and developed pendular nystagmus and esotropia. ... Their unaffected parents were heterozygous for the mutation, which was not found in ethnically matched controls or in the Exome Variant Server. Syndromic Microphthalmia 15 In 96 patients with microphthalmia, Chassaing et al. (2016) analyzed 187 genes associated with ocular development and identified homozygosity for a splice site mutation in the TENM3 gene (610083.0002) in a 9-year-old boy with bilateral colobomatous microphthalmia.
A number sign (#) is used with this entry because of evidence that isolated microphthalmia with coloboma-6 (MCOPCB6) is caused by heterozygous mutation in the GDF3 gene (606522) on chromosome 12p13. For a discussion of genetic heterogeneity of isolated colobomatous microphthalmia, see MCOPCB1 (300345). Clinical Features Ye et al. (2010) reported a mother and daughter with colobomatous microphthalmia. The mother had mild bilateral iris colobomata, mild microphthalmia, normal optic discs and electroretinograms (ERGs), and visual acuity of 20/40 in each eye. The more severely affected daughter exhibited mixed horizontal and rotatory nystagmus, bilateral iris coloboma, severe colobomatous microphthalmia, bilateral foveal hypoplasia, abnormally small optic discs with reduced optic nerve diameters on MRI, visual acuity of 20/200, and abnormal ERGs with decreased a and b wave amplitudes.
She had no findings suggestive of a syndromic malformation. Examination of the father, a brother, and other family members showed no ocular or other physical defects.
A number sign (#) is used with this entry because of evidence that isolated microphthalmia and/or coloboma (MCOPCB10) is caused by heterozygous mutation in the RBP4 gene (180250) on chromosome 10q23. Clinical Features Chou et al. (2015) studied a 7-generation pedigree in which 11 family members had microphthalmia or clinical anophthalmia and/or coloboma. The first proband was an 8-year-old girl with bilateral clinical anophthalmia in whom MRI at day 1 of life showed bilateral absence of the eyeballs, with only cystic remnants in the orbits, thin optic nerves, and a small chiasm; there were no brain abnormalities. The right orbital cyst was surgically removed at 8 months; pathology revealed rudimentary eye structures. The second proband was an 11-year-old boy, a second cousin of the first proband, with left clinical anophthalmia and right microphthalmia with ventronasal iris and chorioretinal coloboma.
Clinically, coloboma is often associated with microphthalmia or clinical anophthalmia and can occur as part of complex malformation syndromes (summary by Wang et al., 2012).
Colobomatous microphthalmia is a developmental disorder of the eye characterized by unilateral or bilateral microphthalmia associated with ocular coloboma.
A number sign (#) is used with this entry because of evidence that isolated colobomatous microphthalmia-7 (MCOPCB7) is caused by heterozygous mutation in the ABCB6 gene (605452) on chromosome 2q35. For a discussion of genetic heterogeneity of isolated colobomatous microphthalmia, see MCOPCB1 (300345). Mapping In members of a 3-generation Chinese family with autosomal dominant iris and chorioretinal coloboma who were negative for mutation in known coloboma-associated genes, Wang et al. (2012) performed genomewide linkage analysis and found linkage to chromosome 2q35. This family was originally reported by Dong et al. (2009). Molecular Genetics In a 3-generation Chinese family with autosomal dominant iris and chorioretinal coloboma mapping to chromosome 2q35 and known to be negative for mutation in known coloboma-associated genes, Wang et al. (2012) sequenced the exons of 76 candidate genes and identified a heterozygous missense mutation in the ABCB6 gene (L811V; 605452.0006) that segregated with disease in the family and was not found in DNA samples from 600 ethnically matched controls. Subsequent analysis of ABCB6 in 116 sporadic Indian coloboma patients, 63 of whom had microphthalmia and coloboma, 21 isolated coloboma, and 32 aniridia, who were all negative for mutation in 9 known coloboma genes, revealed heterozygosity for a different missense mutation (A57T; 605452.0007) in 3 unrelated patients with microphthalmia and coloboma; the mutation was not found in DNA samples from 200 ethnically matched controls.
A number sign (#) is used with this entry because isolated colobomatous microphthalmia-3 (MCOPCB3) is caused by homozygous mutation in the CHX10 gene (142993) on chromosome 14q24. For a discussion of genetic heterogeneity of isolated colobomatous microphthalmia, see MCOPCB1 (300345). Clinical Features Zlotogora et al. (1994) studied isolated colobomatous microphthalmia in multiple relatives of 5 consanguineous families. Microphthalmia was unilateral or bilateral; additional eye findings included microcornea and colobomas of the iris, choroid, optic discs, and/or optic nerve. The intelligence of all the affected members was normal. Three of the families were in an Iranian Jewish community where Zlotogora et al. (1994) suggested that the putative gene may have an unusually high frequency.
For a discussion of genetic heterogeneity of isolated colobomatous microphthalmia, see MCOPCB1 (300345). Clinical Features Morle et al. (2000) studied a 5-generation Sephardic Jewish family in which 7 of 38 members had either unilateral or bilateral microphthalmia of variable severity inherited as an autosomal dominant trait with incomplete penetrance. The inclusion criterion for the diagnosis of microphthalmia was reduction of the total axial length to less than 20 mm in at least 1 eye, determined by ultrasonography. Five of the 7 affected individuals had an ocular prosthesis in at least 1 eye. Two individuals were of uncertain status, since they had ocular anomalies but normal axial length bilaterally.
For a discussion of genetic heterogeneity of isolated microphthalmia with coloboma, see MCOPCB1 (300345). Isolated microphthalmia associated with colobomatous cyst results from a defect in the closure of the embryonic fissure at the 7- to 20-mm stage of development. Microphthalmia can be associated with either a small, clinically undetectable cyst, or a large, typically inferior cyst that deforms the eye and its surroundings. It is usually unilateral, although bilateral cases have been described. Porges et al. (1992) described 5 cases of microphthalmia with colobomatous cyst in 3 separate sibships of a highly inbred kindred.
The 6 factors associated with decreased survival were the development of thrombosis, progression to pancytopenia, myelodysplastic syndrome or acute leukemia, age over 55 years at diagnosis, multiple attempts at treatment, and thrombocytopenia at diagnosis. ... In 7 cases, PNH was associated with aplastic anemia and in 4 with myelodysplastic syndrome. Information on the molecular defect was not provided.
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired clonal hematopoietic stem cell disorder characterized by corpuscular hemolytic anemia, bone marrow failure and frequent thrombotic events. Epidemiology Although PNH has been described worldwide, exact prevalence data are not available. It is estimated at 1/80,000 in France. An incidence estimate of about 1/770,000/year has been reported with a predicted prevalence of approximately 1/62,500 in Great Britain. Higher frequency is suggested in Southeast Asia and in the Far East. Men and women are equally affected. Clinical description The disease may occur at any age but it preferentially affects young adults.
Paroxysmal nocturnal hemoglobinuria is an acquired disorder that leads to the premature death and impaired production of blood cells. The disorder affects red blood cells (erythrocytes), which carry oxygen; white blood cells (leukocytes), which protect the body from infection; and platelets (thrombocytes), which are involved in blood clotting. Paroxysmal nocturnal hemoglobinuria affects both sexes equally, and can occur at any age, although it is most often diagnosed in young adulthood. People with paroxysmal nocturnal hemoglobinuria have sudden, recurring episodes of symptoms (paroxysmal symptoms), which may be triggered by stresses on the body, such as infections or physical exertion. During these episodes, red blood cells are prematurely destroyed (hemolysis).
A number sign (#) is used with this entry because of evidence that susceptibility to paroxysmal nocturnal hemoglobinuria-2 (PNH2) can be conferred by heterozygous mutation in the PIGT gene (610272) on chromosome 20q13. A somatic mutation in the PIGT gene in addition to the germline mutation appears to be necessary for development of the phenotype. One such patient has been reported. For a general phenotypic description and a discussion of genetic heterogeneity of PNH, see PNH1 (300818). Clinical Features Krawitz et al. (2013) reported a Caucasian woman who was diagnosed with hemolytic anemia with a negative direct antiglobulin test at age 44 years. She experienced frequent hemolytic crises, abdominal pain, diarrhea, headache, arthralgia, dyspnea, and fatigue for several year thereafter.
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired disorder that leads to the premature death and impaired production of blood cells. It can occur at any age, but is usually diagnosed in young adulthood. People with PNH have recurring episodes of symptoms due to hemolysis , which may be triggered by stresses on the body such as infections or physical exertion. This results in a deficiency of various types of blood cells and can cause signs and symptoms such as fatigue, weakness, abnormally pale skin (pallor), shortness of breath, and an increased heart rate. People with PNH may also be prone to infections and abnormal blood clotting ( thrombosis ) or hemorrhage, and are at increased risk of developing leukemia .
When this occurs in the foetus , the unmetabolized fatty acids will re-enter the maternal circulation through the placenta, and overwhelm the beta-oxidation enzymes of the mother. [12] The gene responsible for LCHAD has been isolated, and the most common mutation found in acute fatty liver of pregnancy is the E474Q missense mutation . [13] LCHAD deficiency is autosomal recessive in inheritance and mothers are often found to be heterozygous for the affected mutation. [14] Diagnosis [ edit ] The diagnosis of acute fatty liver of pregnancy is usually made on clinical grounds by the treating physician, but differentiation from other conditions affecting the liver may be difficult. [1] The diagnosis of acute fatty liver of pregnancy is suggested by jaundice with a lesser elevation of liver enzymes, elevated white blood cell count, disseminated intravascular coagulation, and a clinically unwell patient. [4] A liver biopsy can provide a definitive diagnosis, [15] but is not always done, due to the increased chance of bleeding in acute fatty liver of pregnancy. [16] Often testing will be done to exclude more common conditions that present in a similar fashion, including viral hepatitis , [17] pre-eclampsia , [5] HELLP syndrome , [4] intrahepatic cholestasis of pregnancy , [1] and autoimmune hepatitis . [3] Pathology [ edit ] If a liver biopsy is needed for diagnosis of the condition, the mother should be appropriately stabilized and treated to reduce bleeding related complications. ... The diagnosis can be enhanced by electron microscopy which can be used to confirm the presence of microvesicular steatosis, and specifically the presence of megamitochondria and paracrystalline inclusions. [18] [19] Liver diseases with similar appearances include Reye's syndrome , drug-induced hepatitis from agents with mitochondrial toxicity, including nucleoside reverse transcriptase inhibitors used to treat HIV , [20] and a rare condition known as Jamaican vomiting sickness which is caused by the eating of the unripened Ackee fruit. [21] Treatment [ edit ] Acute fatty liver of pregnancy is best treated in a centre with expertise in hepatology , high-risk obstetrics , maternal-fetal medicine and neonatology . ... External links [ edit ] Classification D ICD - 10 : O26.6 ICD - 9-CM : 646.7 OMIM : 609016 MeSH : C537957 DiseasesDB : 32879 v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum v t e Pathology of pregnancy , childbirth and the puerperium Pregnancy Pregnancy with abortive outcome Abortion Ectopic pregnancy Abdominal Cervical Interstitial Ovarian Heterotopic Embryo loss Fetal resorption Molar pregnancy Miscarriage Stillbirth Oedema , proteinuria and hypertensive disorders Gestational hypertension Pre-eclampsia HELLP syndrome Eclampsia Other, predominantly related to pregnancy Digestive system Acute fatty liver of pregnancy Gestational diabetes Hepatitis E Hyperemesis gravidarum Intrahepatic cholestasis of pregnancy Integumentary system / dermatoses of pregnancy Gestational pemphigoid Impetigo herpetiformis Intrahepatic cholestasis of pregnancy Linea nigra Prurigo gestationis Pruritic folliculitis of pregnancy Pruritic urticarial papules and plaques of pregnancy (PUPPP) Striae gravidarum Nervous system Chorea gravidarum Blood Gestational thrombocytopenia Pregnancy-induced hypercoagulability Maternal care related to the fetus and amniotic cavity amniotic fluid Oligohydramnios Polyhydramnios Braxton Hicks contractions chorion / amnion Amniotic band syndrome Chorioamnionitis Chorionic hematoma Monoamniotic twins Premature rupture of membranes Obstetrical bleeding Antepartum placenta Circumvallate placenta Monochorionic twins Placenta accreta Placenta praevia Placental abruption Twin-to-twin transfusion syndrome Labor Amniotic fluid embolism Cephalopelvic disproportion Dystocia Shoulder dystocia Fetal distress Locked twins Nuchal cord Obstetrical bleeding Postpartum Pain management during childbirth placenta Placenta accreta Preterm birth Postmature birth Umbilical cord prolapse Uterine inversion Uterine rupture Vasa praevia Puerperal Breastfeeding difficulties Low milk supply Cracked nipples Breast engorgement Childbirth-related posttraumatic stress disorder Diastasis symphysis pubis Postpartum bleeding Peripartum cardiomyopathy Postpartum depression Postpartum psychosis Postpartum thyroiditis Puerperal fever Puerperal mastitis Other Concomitant conditions Diabetes mellitus Systemic lupus erythematosus Thyroid disorders Maternal death Sexual activity during pregnancy Category v t e Inborn error of lipid metabolism : fatty-acid metabolism disorders Synthesis Biotinidase deficiency (BTD) Degradation Acyl transport Carnitine CPT1 CPT2 CDSP CACTD Adrenoleukodystrophy (ALD) Beta oxidation General Acyl CoA dehydrogenase Short-chain SCADD Medium-chain MCADD Long-chain 3-hydroxy LCHAD Very long-chain VLCADD Mitochondrial trifunctional protein deficiency (MTPD): Acute fatty liver of pregnancy Unsaturated 2,4 Dienoyl-CoA reductase deficiency (DECRD) Odd chain Propionic acidemia (PCC deficiency) Other 3-hydroxyacyl-coenzyme A dehydrogenase deficiency (HADHD) Glutaric acidemia type 2 (MADD) To acetyl-CoA Malonic aciduria (MCD) Aldehyde Sjögren–Larsson syndrome (SLS)
A rare, severe complication occurring in the third trimester of pregnancy or in early postpartum period bearing a risk for perinatal and maternal mortality and characterized by jaundice, rise of hepatic injuries and evolving to acute liver failure and encephalopathy.
Acute fatty liver of pregnancy (AFLP) is a rare and serious complication of pregnancy. It is characterized by a build-up of fat in the liver, which can lead to liver damage. The cause of AFLP is not well understood, but genetics may play a role. Symptoms generally begin in the third trimester and may include persistent nausea and vomiting , pain in the stomach or upper-right abdomen, malaise , jaundice and headache. Without prompt treatment, AFLP can lead to coma , organ failure or death of the mother and baby.
However, as noted in this article, the term Potter syndrome was initially coined in order to refer to fetuses and infants with BRA. ... Oligohydramnios sequence with bilateral renal agenesis (Potter's syndrome)" . Journal of Perinatology . 20 (6): 397–8. doi : 10.1038/sj.jp.7200222 . ... "Mermaid and Potter's syndrome occurring simultaneously" (PDF) . ... PMID 20984673 . ^ WELCH RG (May 1958). "The Potter syndrome of renal agenesis" . Br Med J . 1 (5079): 1102–3. doi : 10.1136/bmj.1.5079.1102 . ... "Oligohydramnios, cause of the nonrenal features of Potter's syndrome, including pulmonary hypoplasia".
A number sign (#) is used with this entry because of evidence that renal hypodysplasia/aplasia-2 (RHDA2) is caused by homozygous mutation in the FGF20 gene (605558) on chromosome 8p22. One such family has been reported. Description Renal hypodysplasia/aplasia belongs to a group of perinatally lethal renal diseases, including bilateral renal aplasia, unilateral renal agenesis with contralateral dysplasia (URA/RD), and severe obstructive uropathy. Renal aplasia falls at the most severe end of the spectrum of congenital anomalies of the kidney and urinary tract (CAKUT; 610805), and usually results in death in utero or in the perinatal period. Families have been documented in which bilateral renal agenesis or aplasia coexists with unilateral renal aplasia, renal dysplasia, or renal aplasia with renal dysplasia, suggesting that these conditions may belong to a pathogenic continuum or phenotypic spectrum (summary by Joss et al., 2003; Humbert et al., 2014). For a discussion of genetic heterogeneity of renal hypodysplasia/aplasia, see RHDA1 (191830).
A form of renal agenesis characterized by complete absence of kidney development, absent ureters and subsequent absence of fetal renal function resulting in Potter sequence with pulmonary hypoplasia related to oligohydramnios, which is fatal shortly after birth.
Most had persistent oligohydramnios, severely decreased or absent renal function, and features of Potter syndrome, including dysmorphic facies and clubfeet. ... When BRA was part of a multiple malformation syndrome in a proband, none of the sibs had BRA, although 5 of 40 (12.5%) had a similar pattern of malformations. ... Sanna-Cherchi et al. (2012) concluded that CNV analysis should be performed in individuals with severe renal disorders to make specific syndromic diagnoses and to evaluate for the possibility of later developmental delay. ... Bain et al. (1964) noted that 'Potter syndrome' can be seen in infants with normal kidneys but prolonged leakage of amniotic fluid; Potter syndrome is not pathognomonic of renal anomalies. Scott and Goodburn (1995) found no renal malformations in 50% of autopsied second- or third-trimester fetuses with features of Potter syndrome. There was a high incidence of chorioamnionitis, suggesting that the mechanism of oligohydramnios was occult amniotic fluid leakage.
A rare, congenital renal tract malformation characterized by the complete absence of development of one or both kidneys (unilateral or bilateral renal agenesis respectively), accompanied by absent ureter(s). Epidemiology The birth prevalence of unilateral renal agenesis (RA) is estimated at around 1/2,000. Fetal prevalence of bilateral renal agenesis in Europe has been estimated at 1/8,500. Clinical description Most patients with unilateral RA are asymptomatic early in life if the other kidney is fully functional in which case the condition is commonly detected as an incidental finding later in life. However, hypertension, proteinuria and renal failure may develop in the long run (20-50% of cases at the age of 30).
A number sign (#) is used with this entry because of evidence that renal hypodysplasia/aplasia-3 (RHDA3) is caused by heterozygous mutation in the GREB1L gene (617782) on chromosome 18q11. Description RHDA3 is an autosomal dominant disorder characterized by abnormal kidney development beginning in utero. The phenotype is highly variable, even within families, and there is evidence for incomplete penetrance. Some affected individuals have bilateral renal agenesis, which is usually fatal in utero or in the perinatal period, whereas others may have unilateral agenesis that is compatible with life, or milder manifestations, such as vesicoureteral reflux (VUR). Female mutation carriers may also have uterine or ovarian abnormalities.
There are various causes of Potter sequence including failure of the kidneys to develop ( bilateral renal agenesis ), polycystic kidney diseases , prune belly syndrome, rupture of membranes surrounding the baby, and other kidney abnormalities.
In most nearsighted people, this vision problem is not part of a larger genetic syndrome. However, more than 200 genetic conditions, most of them rare, include nearsightedness as a feature. These conditions include autosomal recessive congenital stationary night blindness, X-linked congenital stationary night blindness, Stickler syndrome, Marfan syndrome, retinitis pigmentosa, cone-rod dystrophy, deafness and myopia syndrome, Knobloch syndrome, and Cohen syndrome. ... When nearsightedness is a feature of a genetic syndrome, it follows the inheritance pattern of that syndrome, most commonly autosomal dominant, autosomal recessive, or X-linked.
Description Myopia, or nearsightedness, is a refractive error of the eye. Light rays from a distant object are focused in front of the retina and those from a near object are focused in the retina; therefore distant objects are blurry and near objects are clear (summary by Kaiser et al., 2004). For a discussion of genetic heterogeneity of myopia, see 160700. Clinical Features Bartsocas and Kastrantas (1981) presented a convincing X-linked pedigree in which 3 myopic brothers had 5 grandsons, through daughters, with myopia. Some of the carrier females had mild myopia ('not requiring corrective glasses'). Although the proband, aged 6.5 years, had short stature, none of the other affected males were short or had hemeralopia or other ocular or physical abnormalities.
Similarities to Wiskott-Aldrich syndrome Cohn et al. (1975) provided follow-up on the kindred of Vestermark and Vestermark (1964). They found evidence of an immunologic defect, thus raising questions of the distinctness of the disorder from Wiskott Aldrich syndrome and from the condition described in entry 314000. ... Molecular Genetics Villa et al. (1995) presented clear evidence that X-linked thrombocytopenia is a disorder allelic to Wiskott-Aldrich syndrome. They found 3 different mutations in the WAS gene in 3 unrelated males with isolated thrombocytopenia and small-sized platelets (300392.0004-300392.0006). None of the 3 patients had other features of the Wiskott-Aldrich syndrome, and none of the 3 mutations had been found in patients with the Wiskott-Aldrich syndrome. ... Wengler et al. (1995) identified 15 novel mutations in patients with full-blown Wiskott-Aldrich syndrome. These mutations involved single basepair changes, or small insertions or deletions, all of which resulted in premature stop codon, frameshift with secondary premature stop codon, or splice site defect.
It is unclear, however, if people with these features have X-linked thrombocytopenia or a more severe disorder with similar signs and symptoms called Wiskott-Aldrich syndrome. Some people have a mild form of the disorder called intermittent thrombocytopenia.
It is a WAS -related disorder, meaning it is caused by a mutation in the Wiskott–Aldrich syndrome ( WAS ) gene, which is located on the short arm of the X chromosome . [1] WAS -related disorders include Wiskott–Aldrich syndrome, XLT, and X-linked congenital neutropenia (XLN). ... This mutation causes the decreased, absent, or altered Wiskott–Aldrich syndrome protein (WASp). Normal WASp is involved in relaying signals from the cell membrane to the actin cytoskeleton . ... Because WAS -related disorders are phenotypically similar, it is important to confirm the absence of the diagnostic criteria for Wiskoff-Aldrich syndrome at the outset. [4] These diagnostic criteria include eczema , lymphoma , autoimmune disorder , recurrent bacterial or viral infections, family history of maternally related males with a WAS -related disorder, and absent or decreased WASp . ... This same research showed that patients with XLT have a high overall survival rate but they are at risk for severe life-threatening complications associated with this disorder, such as serious bleeding events and malignancies. [5] References [ edit ] ^ "WAS - Wiskott-Aldrich syndrome" . Genetics Home Reference . 2014-12-16 .
A 1994 study of 300 healthy blood donors found that 7 persons (2.3%) had FXII deficiencies with one subject having no detectable FXII (0.3%). [5] This study is at variance with estimates that only 1 in 1,000,000 people has the condition. [2] The acquired form of FXII deficiency is seen in patients with the nephrotic syndrome , liver disease, sepsis and shock, disseminated intravascular coagulation , and other diseases. [1] Diagnosis [ edit ] The condition is diagnosed by blood tests in the laboratory when it is noted that special blood clotting test are abnormal. ... External links [ edit ] Classification D ICD - 10 : D68.2 OMIM : 234000 MeSH : D005175 External resources MedlinePlus : 000545 Orphanet : 330 v t e Disorders of bleeding and clotting Coagulation · coagulopathy · Bleeding diathesis Clotting By cause Clotting factors Antithrombin III deficiency Protein C deficiency Activated protein C resistance Protein S deficiency Factor V Leiden Prothrombin G20210A Platelets Sticky platelet syndrome Thrombocytosis Essential thrombocythaemia DIC Purpura fulminans Antiphospholipid syndrome Clots Thrombophilia Thrombus Thrombosis Virchow's triad Trousseau sign of malignancy By site Deep vein thrombosis Bancroft's sign Homans sign Lisker's sign Louvel's sign Lowenberg's sign Peabody's sign Pratt's sign Rose's sign Pulmonary embolism Renal vein thrombosis Bleeding By cause Thrombocytopenia Thrombocytopenic purpura : ITP Evans syndrome TM TTP Upshaw–Schulman syndrome Heparin-induced thrombocytopenia May–Hegglin anomaly Platelet function adhesion Bernard–Soulier syndrome aggregation Glanzmann's thrombasthenia platelet storage pool deficiency Hermansky–Pudlak syndrome Gray platelet syndrome Clotting factor Haemophilia A/VIII B/IX C/XI von Willebrand disease Hypoprothrombinemia/II Factor VII deficiency Factor X deficiency Factor XII deficiency Factor XIII deficiency Dysfibrinogenemia Congenital afibrinogenemia Signs and symptoms Bleeding Bruise Haematoma Petechia Purpura Nonthrombocytopenic purpura By site head Epistaxis Haemoptysis Intracranial haemorrhage Hyphaema Subconjunctival haemorrhage torso Haemothorax Haemopericardium Pulmonary haematoma abdomen Gastrointestinal bleeding Haemobilia Haemoperitoneum Haematocele Haematosalpinx joint Haemarthrosis
A number sign (#) is used with this entry because of evidence that factor XII deficiency is caused by mutation in the F12 gene (610619) on chromosome 5q35. Clinical Features Factor XII deficiency was usually discovered because of the practice in some hospitals of routinely performing whole blood clotting times before surgical operations (McCain et al., 1959). Ratnoff and Steinberg (1962) analyzed data on 55 cases in 37 families. Parental consanguinity was present in at least 2 instances. Some heterozygotes show partial deficiency of Hageman factor. The Japanese case reported by Miwa et al. (1968) had first-cousin parents.
Factor XII deficiency is an inherited disorder that affects a protein (factor XII) involved in blood clotting. While a lack of factor XII does not cause affected individuals to bleed abnormally, the blood takes longer than normal to clot in a test tube. The condition is usually discovered when prolonged clotting is noticed in the process of running other laboratory tests. Factor XII deficiency is caused by mutations in the F12 gene. It is inherited in an autosomal recessive manner.
A rare, autosomal recessive systemic dysfunction of the hemostatic pathway, that is due to a defect in the coagulation factor XII (FXII or Hageman factor), and is either asymptomatic or characterized by a prolonged activated partial thromboplastin time and an increased risk for thromboembolism. FXII deficiency is strongly associated with primary recurrent abortions.
Fetal aortic stenosis is typically detected between 18 and 24 weeks gestation. [2] This early detection is important because it allows for parents to be counseled in a timely and rational manner, allowing for discussion of prognosis and possible outcomes. [2] Another reason for this crucial early detection is because it allows for postnatal management planning. [ citation needed ] Treatment [ edit ] Intervention inutero may need to be done if there is concern that the aortic stenosis is severe enough to lead to the development of hypoplastic left heart syndrome. Management before birth is done by a fetal aortic valvuloplasty. ... Potential open heart surgeries may include aortic valve repair or the Ross procedure . [ citation needed ] If the fetal aortic stenosis is critical it may lead to hypoplastic left heart syndrome . Hypoplastic Left Heart Syndrome (HLHS) it is treated with the Norwood procedure . ... Retrieved from: http://www.childrenshospital.org/health-topics/conditions/aortic-valve-stenosis ^ a b c d e f g Barron, David J; Kilby, Mark D; Davies, Ben; Wright, John GC; Jones, Timothy J; Brawn, William J (2009). "Hypoplastic left heart syndrome". The Lancet . 374 (9689): 551–64. doi : 10.1016/S0140-6736(09)60563-8 . ... "Balloon Dilation of Severe Aortic Stenosis in the Fetus: Potential for Prevention of Hypoplastic Left Heart Syndrome: Candidate Selection, Technique, and Results of Successful Intervention" . ... PMID 2039669 . ^ a b Nationwide Children’s Hospital (2014). Hypoplastic Left Heart Syndrome. Retrieved from: http://www.nationwidechildrens.org/hypoplastic-left-heart-syndrome
A rare aortic malformation of variable severity and clinical presentation. Clinical presentations range from a neonatal severe presentation often associated with sudden cardiac death, to a slowly progressive stenosis that presents later with cardiac murmur, chest pain, dizziness, and loss of consciousness with exercise-induced exacerbations. Echocardiography reveals atresia or dysplasia of the aortic valve most commonly associated with a bicuspid morphology, restricted left ventricular outflow, and left ventricular hypertrophy.
Although arcus may be a manifestation of a disorder of lipid metabolism, it is likely that this is by no means always the case. MacAraeg et al. (1968) showed that arcus corneae occurs in higher frequency and develops at an earlier age in blacks than in whites. They could not relate it to diastolic hypertension, myocardial infarction, or cerebrovascular accidents. Arcus corneae develops precociously in Tangier disease (HDLDT1; 205400), Norum disease, and in homozygotes for type II hyperlipoproteinemia. In osteogenesis imperfecta a ring resembling arcus is seen. The Kayser-Fleischer ring of Wilson disease (277900) bears some similarity.