Scalp–ear–nipple syndrome Other names Finlay-Marks syndrome Scalp–ear–nipple syndrome is inherited in an autosomal dominant manner [1] Scalp–ear–nipple syndrome (also known as "Finlay–Marks syndrome") is a condition associated with aplasia cutis congenita . [2] Contents 1 Presentation 2 Genetics 2.1 Inheritance 3 Diagnosis 4 Treatment 5 Epidemiology 6 Etymology 7 See also 8 References 9 External links Presentation [ edit ] The key affected features of this condition [3] are described in its name. ... Syndrome" by Aase in 1987, [19] “the Finlay Syndrome” by Le Merrer in 1991, [8] the “Scalp-Ear-Nipple Syndrome” by Edwards in 1994, [5] and “Finlay-Marks Syndrome” by Plessis in 1991. [7] The OMIM number OMIM 181270 was assigned in 1987 by Victor A McKusick with the name “Scalp-Ear-Nipple Syndrome” and alternative names “Finlay-Marks Syndrome” and “SEN Syndrome”. [20] See also [ edit ] Say syndrome List of cutaneous conditions References [ edit ] ^ RESERVED, INSERM US14 -- ALL RIGHTS. ... S2CID 1373499 . ^ a b c d Sobreira NL, Brunoni D, Cernach MC, Perez AB. Finlay-Marks (SEN) syndrome: a sporadic case and the delineation of the syndrome. ... "Scalp-ear-nipple (Finlay-Marks) syndrome: a familial case with renal involvement". ... PMID 24660003 . ^ Aase JM (1987). "The Finlay-Marks (S.E.N.) Syndrome: report of a new case and review of the literature".
Scalp-ear-nipple syndrome, as its name suggests, is a condition characterized by abnormalities of the scalp, ears, and nipples. ... The external ears of people with scalp-ear-nipple syndrome may be small, cup-shaped, folded over, or otherwise mildly misshapen. ... Causes Scalp-ear-nipple syndrome is caused by mutations in the KCTD1 gene. ... The mutations in the KCTD1 gene that cause scalp-ear-nipple syndrome impair the transcriptional repressor function of the KCTD1 protein. ... The altered gene activity disrupts normal development of the tissues that arise from the ectoderm (ectodermal dysplasia) and leads to the signs and symptoms of scalp-ear-nipple syndrome. Learn more about the gene associated with Scalp-ear-nipple syndrome KCTD1 Inheritance Pattern Scalp-ear-nipple syndrome is considered to be an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
A number sign (#) is used with this entry because scalp-ear-nipple syndrome (SENS) is caused by heterozygous mutation in the KCTD1 gene (613420) on chromosome 18q11. Description Scalp-ear-nipple syndrome is characterized by aplasia cutis congenita of the scalp, breast anomalies that range from hypothelia or athelia to amastia, and minor anomalies of the external ears. ... The authors suggested that renal and urinary tract abnormalities should be regarded as part of the syndrome. Sobreira et al. (2006) reported a Brazilian boy with Finlay-Marks syndrome. ... Al-Gazali et al. (2007) suggested that the disorder in these children may be a severe recessive form of SEN syndrome. Inheritance Baris et al. (2005) reviewed the literature and concluded that scalp-ear-nipple syndrome is an autosomal dominant condition. Al-Gazali et al. (2007) suggested that there may be a recessive form of SEN syndrome which includes severe hypotonia and developmental delay.
A rare genetic multiple congenital anomalies/dysmorphic syndrome characterized by aplasia cutis congenita of the scalp, breast anomalies ranging from hypothelia or athelia to amastia, and anomalies of the external ears.
These forms of Stickler syndrome are autosomal dominant. Autosomal recessive forms of Stickler syndrome include Stickler syndrome type IV (STL4; 614134), caused by mutation in the COL9A1 gene (120210) on chromosome 6q13, and Stickler syndrome type V (STL5; 614284), caused by mutation in the COL9A2 gene (120260) on chromosome 1p34. ... Kelly et al. (1982) suggested that Weissenbacher-Zweyuller syndrome (WZS; 184840) is a neonatal expression of the Stickler syndrome. ... Clinical Variability Temple (1989) reviewed the clinical variability in Stickler syndrome and discussed phenotypic overlap with Marshall syndrome (154780), Wagner syndrome (143200), and Weissenbacher-Zweymuller syndrome (184840). ... Snead and Yates (1999) provided a review of the clinical and molecular aspects of Stickler syndrome. They discussed the diagnostic features, molecular basis of the condition, and the nosologic difficulties in differentiation from Wagner syndrome, Marshall syndrome, and Weissenbacher-Zweymuller syndrome. ... Genotype/Phenotype Correlations Annunen et al. (1999) identified 15 novel mutations in the COL11A1 gene and 8 in the COL2A1 gene in patients with Marshall syndrome, Stickler syndrome, or Stickler-like syndrome.
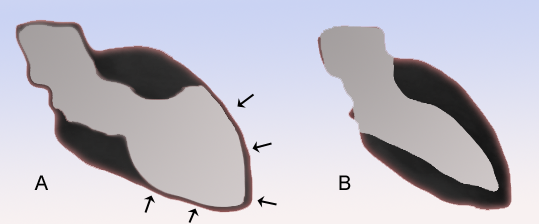
Broken heart syndrome affects just part of the heart. ... Medicines are used to treat symptoms of broken heart syndrome. Broken heart syndrome also may be called: Stress cardiomyopathy. ... Recurrent takotsubo cardiomyopathy. Apical ballooning syndrome. Symptoms Symptoms of broken heart syndrome can mimic a heart attack. ... Risk factors Risk factors for broken heart syndrome include: Sex. Broken heart syndrome is more common in women than in men. ... Blocked arteries do not cause broken heart syndrome. Preparing for your appointment Broken heart syndrome is usually diagnosed in an emergency or hospital setting.
Symptoms are similar to acute coronary syndrome (ACS). Epidemiology Takotsubo syndrome (TTS) is found in about 1-3% of all patients with symptoms of ACS; however, the prevalence is likely underestimated. Around 90% of TTS patients are women, typically postmenopausal but male and younger patients are diagnosed increasingly due to raised awareness of the syndrome. Clinical description In the acute phase, clinical presentation, electrocardiography (ECG) and cardiac biomarkers are similar to those of ACS. ... Etiology The underlying mechanisms of this syndrome remain incompletely understood.
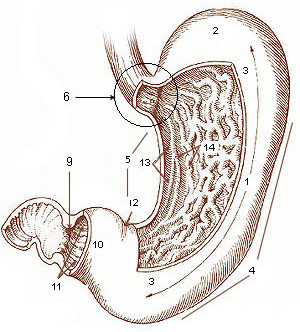
Early dumping syndrome occurs 10 to 30 minutes after a meal. ... Early dumping syndrome symptoms may include: [1] nausea vomiting abdominal pain and cramping diarrhea feeling uncomfortably full or bloated after a meal sweating weakness dizziness flushing, or blushing of the face or skin rapid or irregular heartbeat The symptoms of late dumping syndrome may include: [1] hypoglycemia flushing About 75 percent of people with dumping syndrome report symptoms of early dumping syndrome and about 25 percent report symptoms of late dumping syndrome. Some people have symptoms of both types of dumping syndrome. [1] Diagnosis [ edit ] A health care provider will diagnose dumping syndrome primarily on the basis of symptoms. ... However, surgery to correct dumping syndrome often has unsuccessful results. [1] References [ edit ] ^ a b c d e f g h i j k l m "Dumping Syndrome" . ... (subscription required) ^ "Dumping syndrome - lifestyle and home remedies" .
Sometimes called rapid gastric emptying, dumping syndrome most often occurs as a result of surgery on your stomach or esophagus. ... Generally, you can help prevent dumping syndrome by changing your diet after surgery. ... In more-serious cases of dumping syndrome, you may need medications or surgery. ... And dumping syndrome can develop years after surgery. ... You develop signs and symptoms that might be due to dumping syndrome, even if you haven't had surgery.
Monoamine oxidase A deficiency is a rare disorder that occurs almost exclusively in males. It is characterized by mild intellectual disability and behavioral problems beginning in early childhood. Most boys with monoamine oxidase A deficiency are less able to control their impulses than their peers, causing aggressive or violent outbursts. In addition, affected individuals may have features of other behavioral disorders, including autism spectrum disorder and attention-deficit/hyperactivity disorder (ADHD). These features can include obsessive behaviors, difficulty forming friendships, and problems focusing attention.
A number sign (#) is used with this entry because of evidence that Brunner syndrome (BRNRS) is caused by mutation in the gene encoding monoamine oxidase A (MAOA; 309850) on chromosome Xp11. Description Brunner syndrome is a recessive X-linked disorder characterized by impulsive aggressiveness and mild mental retardation associated with MAOA deficiency (Brunner et al., 1993). ... Palmer et al. (2016) reported 2 unrelated families from Australia with Brunner syndrome. All patients were adults at the time of the report. ... The authors recommended that patients with Brunner syndrome avoid food and medications contraindicated in patients on MAO inhibitors and that they be issued medical alert bracelets. ... In a boy and his 2 maternal uncles with Brunner syndrome, Piton et al. (2014) identified a hemizygous mutation in the MAOA gene (C266F; 309850.0003).
Monoamine oxidase-A deficiency is a very rare recessive X-linked biogenic amine metabolism disorder characterized clinically by mild intellectual deficit, impulsive aggressiveness, and sometimes violent behavior and presenting from childhood.
Monoamine oxidase A deficiency is a rare condition that is characterized by mild intellectual disability and behavioral difficulties (including aggressive and sometimes violent behaviors and autistic features ). Affected people may also experience night terrors, tremor, stereotypical hand movements, and/or occasional body twitches. Signs and symptoms generally develop in childhood and the condition is seen almost exclusively in males. Monoamine oxidase A deficiency is caused by changes (mutations) in the MAOA gene and is inherited in an X-linked recessive manner. Treatment is based on the signs and symptoms present in each person. Some recent studies suggest that cautious treatment with certain medications (called selective serotonin reuptake inhibitors) and dietary modifications can improve symptoms.
Signs and symptoms [ edit ] Buried bumper syndrome may be asymptomatic, especially early in the course. ... The diagnosis is confirmed either endoscopically (via upper endoscopy ) or with computed tomography. [7] Upper endoscopy may reveal overgrowth of stomach tissue over the internal bumper (incomplete buried bumper syndrome). [5] If the bumper has eroded deep into the gastric mucosa, it may not be visualized during endoscopic evaluation (complete buried bumper syndrome). [5] Treatment and prognosis [ edit ] Treatment of buried bumper syndrome consists of removal of the gastrostomy tube. ... Mobilizing and rotating the tube may prevent mucosal overgrowth and aid in avoiding buried bumper syndrome. Severe cases may lead to death. Epidemiology [ edit ] Buried bumper syndrome occurs in 0.3-2.4% of patients. ... The first cases of buried bumper syndrome were reported in 1988 and 1989. [11] The term "buried bumper syndrome" was first used in 1990. [8] See also [ edit ] Percutaneous endoscopic gastrostomy References [ edit ] ^ Pinho, J; Libânio, D; Pimentel-Nunes, P; Dinis-Ribeiro, M (April 2018).
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome. Clinical Features Vincent et al. (1994) analyzed a de novo 8q12.2-q21.2 deletion with the identification of a proposed 'new' contiguous gene syndrome consisting of the branchiootorenal (BOR) syndrome (113650), Duane syndrome (126800), hydrocephalus (600256), and aplasia of the trapezius muscle. This was the first reported localization of the genes responsible for Duane syndrome and for a dominant form of hydrocephalus. ... Using new algorithms, Vincent et al. (1994) constructed a YAC contig and used it to localize the breakpoint of another chromosomal rearrangement associated with the branchiootic syndrome to a 500-kb interval within the deletion. Eyes - Duane syndrome Neuro - Hydrocephalus Inheritance - Autosomal dominant contiguous gene syndrome Lab - Deletion of 8q12.2-q21.2 Muscle - Trapezius muscle aplasia Ears - Branchiootorenal (BOR) syndrome ▲ Close
Clinical Features The Wildervanck syndrome consists of congenital perceptive deafness, Klippel-Feil anomaly (see 118100), and abducens palsy with retractio bulbi (Duane syndrome). ... Kirkham (1969) described a family which was affected through 5 generations with perceptive deafness and in which 2 members had Duane syndrome. Konigsmark and Gorlin (1976) favored multifactorial inheritance. ... The authors suggested that children with Wildervanck syndrome should be investigated for craniospinal abnormalities by MRI. Abu-Amero et al. (2014) described a male with Wildervanck syndrome who had a microdeletion in the X chromosome. ... Cytogenetics In a male patient with Wildervanck syndrome, Abu-Amero et al. (2014) detected an approximately 3-kb deletion in chromosome Xq26.3 (chrX:137,779,548-137,782,146, NCBI35) using array CGH.
Wildervanck syndrome is a condition that affects the bones in the neck, the eyes, and the ears. It is characterized by Klippel-Feil anomaly (in which the bones of the neck fuse together), Duane syndrome (an eye movement disorder), and hearing loss. Wildervanck syndrome occurs primarily in females. In most cases, Wildervanck syndrome occurs randomly for unknown reasons in a family with no prior history (sporadically), though a deletion on the X chromosome was identified in one individual with Wildervanck syndrome.
Wildervanck syndrome is characterized by the triad of cervical vertebral fusion (Klippel-Feil anomaly, see this term), bilateral abducens palsy with retracted eyes (Duane syndrome, see this term) and congenital perceptive deafness.
A number sign (#) is used with this entry because this disorder, which comprises seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SESAME syndrome), is caused by homozygous or compound heterozygous mutation in the KCNJ10 gene (602208) on chromosome 1q23. ... Scholl et al. (2009) termed this disorder 'SeSAME syndrome.' Bockenhauer et al. (2009) reported a consanguineous family of Pakistani origin in which 4 children presented in infancy with generalized tonic-clonic seizures. ... Bockenhauer et al. (2009) referred to the disorder as 'EAST syndrome' for epilepsy, ataxia, sensorineural deafness, and tubulopathy. ... In affected members of a Pakistani family with EAST syndrome, Bockenhauer et al. (2009) identified a homozygous mutation in the KCNJ10 gene (602208.0001). In 6 patients from 4 families with EAST syndrome, Freudenthal et al. (2011) identified 4 different homozygous mutations in the KCNJ10 gene (602208.0006, 602208.0010-602208.0012).
SeSAME syndrome is characterized by Sei zures , S ensorineural deafness , A taxia (lack of muscle coordination), intellectual ( M ental) disability , and E lectrolyte imbalance (low levels of potassium and magnesium in the blood, hypokalemia and hypomagnesemia, and metabolic alkalosis ). It may also be known as EAST syndrome ( E pilepsy , A taxia, S ensorineural deafness, and T ubulopathy ( kidney problems in the structures known as tubules)). ... The ataxia seems to be the most debilitating feature of the syndrome. It is caused by mutations in the KCNJ10 gene, and inherited in an autosomal recessive pattern.
SeSAME syndrome is characterized by seizures, sensorineural deafness, ataxia, intellectual deficit, and electrolyte imbalance (hypokalemia, metabolic alkalosis, and hypomagnesemia).
Mirhosseini–Holmes–Walton syndrome Other names Pigmentary retinopathy-intellectual disability syndrome Specialty Ophthalmology, Neurology Mirhosseini–Holmes–Walton syndrome is a syndrome which involves retinal degeneration , cataract , microcephaly , and mental retardation . It was first characterized in 1972. [1] [2] There is evidence that this syndrome has a different mutation in the same gene as Cohen syndrome . [3] [4] References [ edit ] ^ Mirhosseini, SA; Holmes, LB; Walton, DS (1972). "Syndrome of pigmentary retinal degeneration, cataract, microcephaly, and severe mental retardation" . ... "Tapetoretinal degeneration in brothers with apparent Cohen syndrome: Nosology with Mirhosseini-Holmes-Walton syndrome". ... "Are the Mirhosseini-Holmes-Walton syndrome and the Cohen syndrome identical?".
Horn et al. (2000) reported 2 brothers and a cousin from a multiply consanguineous kindred of Lebanese descent with a syndrome of microcephaly, progressive postnatal growth deficiency, mental retardation, hypotonia, chorioretinal dystrophy, and myopia. ... This region overlaps the refined gene region for Cohen syndrome (COH1; 216550). Horn et al. (2000) hypothesized that the syndrome in their family, Cohen syndrome, and Mirhosseini-Holmes-Walton syndrome may be allelic. INHERITANCE - Autosomal recessive GROWTH Height - Short stature, moderate Weight - Truncal obesity HEAD & NECK Head - Microcephaly Eyes - Retinal pigmentary degeneration - Myopia - Decreased visual acuity Teeth - Prominent incisors SKELETAL Limbs - Hyperextensible joints Hands - Narrow hands - Arachnodactyly Feet - Narrow feet NEUROLOGIC Central Nervous System - Severe mental retardation MISCELLANEOUS - Possibly allelic to Cohen syndrome ( 216550 ) ▲ Close
Congenital intrauterine infection-like syndrome is characterised by the presence of microcephaly and intracranial calcifications at birth accompanied by neurological delay, seizures and a clinical course similar to that seen in patients after intrauterine infection with Toxoplasma gondii, Rubella, Cytomegalovirus, Herpes simplex (so-called TORCH syndrome), or other agents, despite repeated tests revealing the absence of any known infectious agent. ... Differential diagnosis Congenital intrauterine infection-like syndrome shows considerable clinical overlap with Aicardi-Goutieres syndrome (AGS, see this term). The two syndromes were reported to differ by the presence of cerebrospinal fluid anomalies (CSF leucocytosis and elevated IFN-alpha levels) in AGS and hepatic dysfunction, congenital microcephaly and thrombocytopaenia in congenital intrauterine infection-like syndrome. However, as the clinical manifestations of both syndromes show significant variability, it has been suggested that AGS and congenital intrauterine infection-like syndrome represent different presentations of the same disease. The differential diagnosis should also include other syndromes characterised by microcephaly and intracranial calcification such as Cockayne syndrome, COFS syndrome (which is usely considered as the neonatal form of Cockayne syndrome) and Hoyeraal-Hreidarsson syndrome (the neonatal presentation of dyskeratosis congenita syndrome; see these terms), some cases of mitochondrial encephalomyopathy, and pseudohypoarathyroidism.
Schmitt et al. (1982) described a family in which 5 females and 3 males over 3 generations had bilateral, symmetric, nonopposable triphalangeal thumbs and radial hypoplasia. Affected males had first-degree hypospadias and all affected persons had anterior maxillary diastema. No male-to-male transmission was observed; however, transmission to only 2 of 4 daughters by an affected male favors autosomal (as opposed to X-linked) dominant inheritance. Limbs - Nonopposable triphalangeal thumbs - Radial hypoplasia Inheritance - Autosomal dominant GU - First-degree hypospadias in males Facies - Anterior maxillary diastema ▲ Close
Radial hypoplasia-triphalangeal thumbs-hypospadias-maxillary diastema syndrome is characterised by symmetric, nonopposable triphalangeal thumbs and radial hypoplasia. ... The affected males also presented with hypospadias. The syndrome is inherited as an autosomal dominant trait.
Syndrome characterized by acute brain damage and liver function problems Reye syndrome Other names Reye's syndrome Appearance of a liver from a child who died of Reye syndrome as seen with a microscope . ... Eight of the nine children with Reye syndrome were found to have been exposed to aspirin. ... Also in 1964, George Johnson and colleagues published an investigation of an outbreak of influenza B that described 16 children who developed neurological problems, four of whom had a profile remarkably similar to Reye syndrome. Some investigators refer to this disorder as Reye-Johnson syndrome, although it is more commonly called Reye syndrome. ... Archived from the original on March 6, 2013. ^ "What is Reye's Syndrome?" . National Reye's Syndrome Foundation. 1974. ... PMID 2680560 . ^ Gosalakkal JA, Kamoji V (September 2008). "Reye syndrome and reye-like syndrome". Pediatric Neurology . 39 (3): 198–200. doi : 10.1016/j.pediatrneurol.2008.06.003 .
Overview Reye's syndrome, also known as Reye syndrome, is a rare but serious condition that causes swelling in the liver and brain. ... When to see a doctor Early diagnosis and treatment of Reye's syndrome can save a child's life. If you suspect that your child has Reye's syndrome, it's important to act quickly. ... Causes The use of aspirin during a viral illness has most commonly been linked to Reye's syndrome. But the exact cause of Reye's syndrome is unknown. ... Reye's syndrome may develop after influenza or chickenpox in particular. ... Avoiding these two viral illnesses can help prevent Reye's syndrome. Diagnosis There's no specific test for Reye's syndrome.
A rare, systemic disease characterized by persistent vomiting with confusion, lethargy, disorientation, hyperreflexia, hyperventilation, and tachycardia, with rapid progression to seizures, non-inflammatory encephalopathy, coma and death. It typically develops between 12 hours and 3 weeks after recovery from a viral illness, such as upper respiratory tract infection or gastroenteritis. Hepatomegaly, acute hepatic steatosis, fatty liver degeneration and multiple laboratory abnormalities are associated.
Silver syndrome belongs to a group of genetic disorders known as hereditary spastic paraplegias. ... The first sign of Silver syndrome is usually weakness in the muscles of the hands. ... Frequency Although Silver syndrome appears to be a rare condition, its exact prevalence is unknown. ... Some people with Silver syndrome do not have an identified mutation in the BSCL2 gene. ... Learn more about the gene associated with Silver syndrome BSCL2 Inheritance Pattern Silver syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
The authors noted the variation in the clinical phenotype, which had features of distal spinal muscular atrophy, pure pyramidal syndromes, and peroneal muscular atrophy with pyramidal features. ... Brusse et al. (2009) reported 12 members of a large 3-generation Dutch family with phenotypic overlap between Silver syndrome and distal HMN5 who carried a heterozygous N88S mutation. ... Mapping In 2 large multigeneration families with Silver syndrome, 1 of which had been reported by Silver (1966), Patel et al. (2001) excluded linkage to known loci for spastic paraplegia. ... In 4 related Austrian families with typical Silver syndrome, Windpassinger et al. (2003) found linkage to 11q12-q14. ... In affected members of a Belgian family and a Brazilian family with Silver syndrome, Windpassinger et al. (2004) identified heterozygosity for a ser90-to-leu mutation (S90L; 606158.0014).
A complex hereditary spastic paraplegia characterized by progressive spastic paraplegia, upper and lower limb muscle atrophy, hyperreflexia, extensor plantar responses, pes cavus and occasionally impaired vibration sense. Association with hand muscles amyotrophy typical.
Kennedy's disease, also known as bulbospinal muscular atrophy (BSMA), is a rare X-linked recessive motor neuron disease characterized by proximal and bulbar muscle wasting. Epidemiology The prevalence of BSMA is 1/30,000 male births. The incidence is 1/526,315 males/year. Clinical description Disease onset occurs between 30-60 years of age. Initial clinical manifestations include tremor, muscle cramps, muscle twitching, fatigue and slurred speech. With disease progression patients additionally develop weakness and wasting of the limb and bulbar muscles, manifesting as dysarthria, dysphonia, hanging jaw, tongue wasting, chewing difficulty and impaired mobility.
Women with MRKH syndrome have a female chromosome pattern (46,XX) and normally functioning ovaries. ... When only reproductive organs are affected, the condition is classified as MRKH syndrome type 1. Some women with MRKH syndrome also have abnormalities in other parts of the body; in these cases, the condition is classified as MRKH syndrome type 2. ... Frequency MRKH syndrome affects approximately 1 in 4,500 newborn girls. Causes The cause of MRKH syndrome is unknown. Changes in several genes that are involved in development before birth have been identified in females with MRKH syndrome. ... Less often, MRKH syndrome is passed through generations in families.
The abnormality in sexual development in this syndrome was similar to that seen in the Rokitansky-Kuster-Hauser syndrome (277000). ... Guerrier et al. (2006) reviewed the clinical features of the MRKH syndrome and MURCS association phenotypes and discussed genetic hypotheses. Noting that combinations of wolffian duct agenesis or severe hypoplasia with or without renal and/or skeletal anomalies have been described, the authors suggested that the term GRES syndrome (for genital, renal, ear, and skeletal) might be more appropriate when applied to both sexes.
It can be classified as either MRKH syndrome type 1 (corresponding to isolated utero-vaginal aplasia) or MRKH syndrome type 2 (utero-vaginal aplasia associated with other malformations) (see these terms). Epidemiology MRKH syndrome has a worldwide incidence of 1/4500 live female births. ... MRKH syndrome type 1 and 2 patients lack the uterus and the upper 2/3 of the vagina leading to difficulties with sexual intercourse in some. ... Differential diagnosis Differential diagnosis includes isolated vaginal atresia, which is found in various syndromes such as McKusick-Kaufman syndrome, androgen insensitivity syndrome, Mullerian aplasia and hyperandrogenism, and renal-genital-middle ear anomalies (see these terms). ... For the associated malformations in MRKH type 2, specific medical care is directed toward the anomalies. Prognosis MRKH syndrome is not a life threatening disease.
The underlying causes of MRKH syndrome is still being investigated, but several causative genes have been studied for their possible association with the syndrome. ... "Mayer–Rokitansky–Kuster–Hauser syndrome: recent clinical and genetic findings". ... "Modernes Management der angeborenen (Mayer-Rokitansky-Küster-Hauser, MRKH-Syndrom) und erworbenen Vaginalaplasie" (PDF) . ... "Evaluation and management of cases of primary amenorrhoea with MRKH syndrome" . Bangladesh Medical Journal Khulna . 45 (1–2): 24–29. doi : 10.3329/bmjk.v45i1-2.13626 . ^ "Rokitansky Syndrome: Information for Parents / Carers" (PDF) . ... "Prevalence and patient characteristics of Mayer–Rokitansky–Küster–Hauser syndrome: a nationwide registry-based study" .