-
Idiopathic Guttate Hypomelanosis
Wikipedia
External links [ edit ] Classification D ICD - 10 : L81.5 ( ILDS L81.540) DiseasesDB : 31365 External resources eMedicine : article/1068422 External links [ edit ] v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation This cutaneous condition article is a stub .
-
Lichen Aureus
Wikipedia
External links [ edit ] Classification D DiseasesDB : 31388 v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation This cutaneous condition article is a stub .
-
Björnstad Syndrome
Wikipedia
Björnstad syndrome Other names BJS [1] Björnstad syndrome is an autosomal recessive congenital condition involving pili torti , [2] nerve deafness and hair abnormalities. ... It is caused by mutations in the BCS1L gene which also cause GRACILE syndrome. [6] See also [ edit ] GRACILE syndrome References [ edit ] ^ "OMIM Entry - # 262000 - Bjornstad Syndrome; BJS" . omim.org . ... "Missense mutations in the BCS1L gene as a cause of the Björnstad syndrome". N. Engl. J. Med . 356 (8): 809–19. doi : 10.1056/NEJMoa055262 . ... ISSN 0028-4793 . PMID 17314340 . ^ "Bjornstad syndrome" . NCATS . Genetic and rare diseases information center . ... External links [ edit ] Classification D OMIM : 262000 MeSH : C537633 DiseasesDB : 33516 External resources Orphanet : 123 v t e Congenital malformations and deformations of skin appendages Nail disease Anonychia Leukonychia Pachyonychia congenita / Onychauxis Koilonychia Hair disease hypotrichosis /abnormalities: keratin disease Monilethrix IBIDS syndrome Sabinas brittle hair syndrome Pili annulati Pili torti Uncombable hair syndrome Björnstad syndrome Giant axonal neuropathy with curly hair hypertrichosis : Zimmermann–Laband syndrome v t e Disorders of citric acid cycle and electron transport chain Citric acid cycle Pyruvate dehydrogenase deficiency Fumarase deficiency Electron transport chain Coenzyme Q10 deficiency Björnstad syndrome GRACILE syndrome Leigh's disease This genetic disorder article is a stub .
-
Weill-Marchesani Syndrome
Medlineplus
Weill-Marchesani syndrome is a disorder of connective tissue. ... Adult height for men with Weill-Marchesani syndrome ranges from 4 feet, 8 inches to 5 feet, 6 inches. ... Occasionally, heart defects or an abnormal heart rhythm can occur in people with Weill-Marchesani syndrome. Frequency Weill-Marchesani syndrome appears to be rare; it has an estimated prevalence of 1 in 100,000 people. ... Mutations in this gene disrupt the normal development of these structures, which leads to the specific features of Weill-Marchesani syndrome. A mutation in the FBN1 gene has also been found to cause Weill-Marchesani syndrome. ... Learn more about the genes associated with Weill-Marchesani syndrome ADAMTS10 FBN1 Inheritance Pattern Weill-Marchesani syndrome can be inherited in either an autosomal recessive or an autosomal dominant pattern.
-
Riehl Melanosis
Wikipedia
Chichester, UK: John Wiley & Sons. p. 37. v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation This cutaneous condition article is a stub .
-
Megdel Syndrome
Medlineplus
MEGDEL syndrome is an inherited disorder that affects multiple body systems. ... People with MEGDEL syndrome also have high urine levels of another acid called 3-methylglutaric acid. ... Another feature of MEGDEL syndrome is brain dysfunction (encephalopathy). ... People with MEGDEL syndrome have changes in the brain that resemble those in another condition called Leigh syndrome. ... Frequency MEGDEL syndrome is a rare disorder; its prevalence is unknown.
-
Dyschromia
Wikipedia
External links [ edit ] Classification D ICD - 10 : L81.9 ICD - 9-CM : 709.0 DiseasesDB : 29542 SNOMED CT : 201273003 v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation This cutaneous condition article is a stub .
-
Periorbital Hyperpigmentation
Wikipedia
External links [ edit ] Classification D ICD - 10 : L81.4 ( ILDS L81.408) v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation This cutaneous condition article is a stub .
-
Nonsyndromic Deafness
Wikipedia
In contrast, syndromic deafness involves hearing loss that occurs with abnormalities in other parts of the body. ... The relative contribution of heredity to age-related hearing impairment is not known, however the majority of inherited late-onset deafness is autosomal dominant and non-syndromic (Van Camp et al., 1997). Over forty genes associated with autosomal dominant non-syndromic hearing loss have been localized and of these fifteen have been cloned. ... Different mutations in the same gene can cause different types of hearing loss, and some genes are associated with both syndromic and nonsyndromic deafness. In many families , the gene(s) involved have yet to be identified. ... Most cases of genetic deafness (70% to 80%) are nonsyndromic; the remaining cases are caused by specific genetic syndromes. In adults, the chance of developing hearing loss increases with age; hearing loss affects half of all people older than 80 years. ... In GeneReviews External links [ edit ] Classification D ICD - 10 : H90.5 MeSH : C580334 External resources Orphanet : 87884 v t e Disorders of hearing and balance Hearing Symptoms Hearing loss Excessive response Tinnitus Hyperacusis Phonophobia Disease Loss Conductive hearing loss Otosclerosis Superior canal dehiscence Sensorineural hearing loss Presbycusis Cortical deafness Nonsyndromic deafness Other Deafblindness Wolfram syndrome Usher syndrome Auditory processing disorder Spatial hearing loss Tests Hearing test Rinne test Tone decay test Weber test Audiometry pure tone visual reinforcement Balance Symptoms Vertigo nystagmus Disease Balance disorder Peripheral Ménière's disease Benign paroxysmal positional vertigo Labyrinthitis Labyrinthine fistula Tests Dix–Hallpike test Unterberger test Romberg's test Vestibulo–ocular reflex v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions ) v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins v t e Genetic disorder , membrane: Solute carrier disorders 1-10 SLC1A3 Episodic ataxia 6 SLC2A1 De Vivo disease SLC2A5 Fructose malabsorption SLC2A10 Arterial tortuosity syndrome SLC3A1 Cystinuria SLC4A1 Hereditary spherocytosis 4 / Hereditary elliptocytosis 4 SLC4A11 Congenital endothelial dystrophy type 2 Fuchs' dystrophy 4 SLC5A1 Glucose-galactose malabsorption SLC5A2 Renal glycosuria SLC5A5 Thyroid dyshormonogenesis type 1 SLC6A19 Hartnup disease SLC7A7 Lysinuric protein intolerance SLC7A9 Cystinuria 11-20 SLC11A1 Crohn's disease SLC12A3 Gitelman syndrome SLC16A1 HHF7 SLC16A2 Allan–Herndon–Dudley syndrome SLC17A5 Salla disease SLC17A8 DFNA25 21-40 SLC26A2 Multiple epiphyseal dysplasia 4 Achondrogenesis type 1B Recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia SLC26A4 Pendred syndrome SLC35C1 CDOG 2C SLC39A4 Acrodermatitis enteropathica SLC40A1 African iron overload see also solute carrier familyGJB3, MYO15A, PCDH15, DCDC2, TMC1, MYO7A, COL11A2, KCNQ4, CDH23, TMPRSS3, MYO6, POU4F3, GIPC3, CIB2, TBC1D24, ILDR1, POU3F4, LHFPL5, COCH, TPRN, USH1C, MARVELD2, TECTA, ACTG1, GRXCR1, PJVK, CDC14A, MYH14, OSBPL2, WHRN, P2RX2, TMEM132E, SMPX, TMIE, KARS1, CEACAM16, EYA4, GJA1, STRC, SLC17A8, CABP2, CD164, TJP2, TNC, ESPN, RDX, ELMOD3, MYO3A, OTOGL, CEMIP, LOXHD1, ESRRB, NARS2, EPS8L2, MET, HOMER2, MSRB3, RIPOR2, OTOG, SLC26A5, DIABLO, BDP1, KITLG, TGFA, GRHL2, ROR1, OTOA, CCDC50, MSX1, MYO1F, CLDN14, TMTC2, USH1G, SYNE4, GRXCR2, SLC44A4, SERPINB6, HGF, ADGRV1, TGFB3, TSPEAR, ALG10, PPIP5K2, GJB6, OTOF, SMIM12, GJB2, SLC26A4, GJC3, GSDME, RNR1, TRIOBP, LRTOMT, PRPS1, WFS1, ACTB, PTGDS, TRNS1, CIB1, DIAPH1, UCN, USH2A, MPZL2, GJB4, GPSM2, CFAP410, DFNB13, MITF, MCM2, LRP5, LMX1A, KCNJ10, KCNE1, DFNA21, GJB1, FER, DFNA59, DFNB14, DFNX3, COL4A6, DFNB65, TIMM8A, AIRE, CDAN1, DFNB38, CRYM, MAP3K8, PTPRQ, GJB7, ASCL1, DFNB46, COL11A1, CSTB, DFNA24, ABCC1, BSND, HARS2, TRS-AGA2-3, DMXL2, CLIC5, MYH15, GRAP, RAI1, ASH1L, LHFPL6, GJC1, CLDN9, MLRL, DNALI1, COX1, PDZD7, TOP2B, TCF3, SULT1E1, SIX1, PNPT1, PDE1C, NLRP3, MYO1A, TMC2, MYH9, DFNA30, DFNB96
-
Renpenning Syndrome
Orphanet
Renpenning syndrome is an X-linked intellectual disability syndrome (XLMR, see this term) characterized by intellectual deficiency, microcephaly, leanness and mild short stature. ... Phenotypic variants grouped under Renpenning syndrome include Golabi-Ito-Hall syndrome, Hamel cerebro-palato-cardiac syndrome (see these terms), Porteous syndrome, Sutherland-Haan syndrome, MRX55 and three other XLMR families. Hamel cerebro-palato-cardiac syndrome usually has the most severe manifestations. ... Differential diagnosis Fragile X syndrome is a differential diagnosis but microcephaly is not a feature. Other causes of microcephaly such as fetal CMV infection, fetal alcohol syndrome, maternal phenylketonuria, autosomal recessive microcephalies and Smith-Lemli-Opitz syndrome (see this term) should be considered.PQBP1, STS, MAPK1, RNF19A, SIRT1, AHSA1, GRAP2, AIMP2, TNF, IL1B, RUNX2, HSP90AA1, GSTM2, G6PD, FMR1, DLG3, MAPK14, CRK, POLDIP2

-
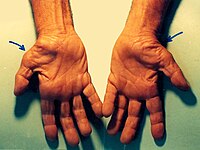
Carpal Tunnel Syndrome
Medlineplus
While carpal tunnel syndrome can occur at any age, it most often affects people between the ages of 40 and 60. ... Frequency Carpal tunnel syndrome is estimated to affect 1 to 5 percent of the adult population. ... Learn more about the genes associated with Carpal tunnel syndrome COL11A1 COL1A1 COL5A1 TTR Additional Information from NCBI Gene: BGN GSTM1 IL6R SH3TC2 Inheritance Pattern Carpal tunnel syndrome is a complex condition and is usually not inherited. However, having a close relative with carpal tunnel syndrome likely increases a person's risk of developing the condition. When carpal tunnel syndrome occurs as part of a genetic syndrome, this feature follows the inheritance pattern of the syndrome.TTR, AOC1, ADAMTS10, AEBP1, SMAD6, LTBP1, EFEMP1, HCP5, TTLL5, UMPS, ADAMTS17, B2M, PMP22, CCN2, IL1B, PSMD9, CYP19A1, GSTT1, COL5A1, PGR, SLX1A-SULT1A3, SEMG1, PDGFRB, SULT1A4, MIR106A, MAPK3, PDGFRA, SERPINE1, PTGS2, ROS1, CCL2, DHDDS, SLC6A2, SULT1A3, CHPT1, TGFB1, TGFBR1, GSTK1, NT5E, VDR, VEGFA, HDAC4, ADAMTS5, SLCO6A1, INTU, HPGDS, SEMA5B, OLR1, MMP3, NDUFB2, CECR, ELK3, ACE, CSH2, CSH1, COMT, COL11A1, ERCC8, CASP8, COX2, CALCR, BGN, AMH, ABCD1, ALB, AKT1, ACAN, EPHB1, ESR1, FAP, GHR, MPZ, MMP10, MMP9, MMP1, MIP, IL6R, IL1RN, IGF1, TNC, HSPA9, HLA-DRB3, HLA-DPB1, HLA-DPA1, GSTP1, GSTM1, MTCO2P12
-
Antiphospholipid Syndrome
Medlineplus
Women with antiphospholipid syndrome are at increased risk of complications during pregnancy. ... At birth, infants of mothers with antiphospholipid syndrome may be small and underweight. A thrombosis or pregnancy complication is typically the first sign of antiphospholipid syndrome. ... Less than 1 percent of individuals with antiphospholipid syndrome develop CAPS. Frequency The exact prevalence of antiphospholipid syndrome is unknown. ... It is estimated that 20 percent of individuals younger than age 50 who have a stroke have antiphospholipid syndrome. Ten to 15 percent of people with systemic lupus erythematosus have antiphospholipid syndrome. ... Approximately 70 percent of individuals diagnosed with antiphospholipid syndrome are female. Causes The genetic cause of antiphospholipid syndrome is unknown.
-
Revesz Syndrome
Wikipedia
Revesz syndrome Other names Dyskeratosis congenita with bilateral exudative retinopathy [1] Revesz syndrome is inherited in an autosomal dominant manner Causes Genetic Revesz syndrome is a fatal disease that causes exudative retinopathy and bone marrow failure. [2] Other symptoms include severe aplastic anemia , intrauterine growth retardation, fine sparse hair, fine reticulate skin pigmentation, [3] ataxia due to cerebellar hypoplasia , and cerebral calcifications . ... Patients with Revesz syndrome have presented with heterozygous mutations in TINF2 gene which is located on chromosome 14q12. ... You can help by adding to it . ( October 2017 ) Epidemiology [ edit ] Revesz syndrome has so far been observed only in children. ... "Revesz syndrome". Indian Journal of Pediatrics . 74 (9): 862–863. doi : 10.1007/s12098-007-0155-2 . ... External links [ edit ] Classification D OMIM : 268130 MeSH : C538371 DiseasesDB : 34808 External resources Orphanet : 3088 v t e Nucleus diseases Telomere Revesz syndrome Nucleolus Treacher Collins syndrome Spinocerebellar ataxia 7 Cajal body : Spinal muscular atrophy Centromere CENPJ Seckel syndrome 4 Other AAAS Triple-A syndrome Laminopathy SMC1A / SMC3 Cornelia de Lange Syndrome SETBP1 Schinzel–Giedion syndrome see also nucleus

-
48,xxxy Syndrome
Medlineplus
However, some boys and men with 48,XXXY syndrome do not have these differences in their facial features. 48,XXXY syndrome disrupts male sexual development. ... Boys and men with 48,XXXY syndrome have extra copies of multiple genes on the X chromosome. ... Researchers are working to determine which genes contribute to the specific developmental and physical differences that occur with 48,XXXY syndrome. 48,XXXY syndrome is sometimes described as a variant of another sex chromosome disorder called Klinefelter syndrome. ... Like 48,XXXY syndrome, Klinefelter syndrome affects male sexual development and can be associated with learning disabilities and problems with speech and language development. However, the features of 48,XXXY syndrome tend to be more severe than those of Klinefelter syndrome and affect more parts of the body.
-
Berdon Syndrome
Wikipedia
Berdon syndrome Other names Megacystis-microcolon-intestinal hypoperistalsis syndrome, MMIH syndrome, MMIHS Berdon syndrome has an autosomal recessive pattern of inheritance . Specialty Medical genetics Berdon syndrome , also called Megacystis-microcolon-intestinal hypoperistalsis syndrome ( MMIH syndrome ), [1] is an autosomal recessive [2] fatal [3] genetic disorder affecting newborns. ... "Megacystis microcolon intestinal hypoperistalsis syndrome: Systematic review of outcome". ... "Megacystis-microcolon-intestinal hypoperistalsis syndrome: A new cause of intestinal obstruction in the newborn. ... External links [ edit ] Megacystis microcolon intestinal hypoperistalsis syndrome at NIH 's Office of Rare Diseases Online Mendelian Inheritance in Man (OMIM): Megacystis microcolon intestinal hypoperistalsis syndrome; MMIH syndrome; Berdon syndrome - 249210 Classification D OMIM : 249210 MeSH : C536138 DiseasesDB : 32131

-
Rab18 Deficiency
Medlineplus
Individuals with Martsolf syndrome have cataracts, microphthalmia, and small pupils. They have milder optic atrophy and cortical visual impairment than people with Warburg micro syndrome. Intellectual disability is mild to moderate in people with Martsolf syndrome. ... Hypotonia is common in infants with Martsolf syndrome, although spasticity worsens more slowly than in individuals with Warburg micro syndrome, and it usually affects only the legs and feet. Hypogonadotropic hypogonadism can also occur in individuals with Martsolf syndrome. Neither Warburg micro syndrome nor Martsolf syndrome affect the life expectancy of affected individuals. Frequency RAB18 deficiency is rare; its exact prevalence is unknown. Warburg micro syndrome is more common than Martsolf syndrome.
-
Gilbert Syndrome
Mayo_clinic
Gilbert syndrome requires no treatment. Symptoms The most frequent sign of Gilbert syndrome is an occasional yellowish tinge of the skin and the whites of the eyes as a result of slightly higher levels of bilirubin in the blood. ... Causes Gilbert syndrome is caused by a modified gene you inherit from your parents. ... Should I have my bilirubin level tested again? Could Gilbert syndrome cause my signs and symptoms? Could the medications I'm taking for other conditions worsen Gilbert syndrome? Can Gilbert syndrome cause complications or lead to liver damage? ... How likely is it that my children will inherit Gilbert syndrome?UGT1A1, UGT1A, UGT1A10, UGT1A8, UGT1A7, UGT1A6, UGT1A4, UGT1A3, UGT1A5, UGT1A9, SLC35A2, G6PD, UGGT1, HFE, CNDP2, CHPT1, ABCG5, DHDDS, NT5C3A, NT5C2, UROD, NAT2, ABO, SLC10A2, ACP3, ABCC2, CYP2D7, CYP2D6, DPYD, GPT, IBSP, IL1B, IL6, NT5E, PPARA, PRKAA1, PRKAA2, PRKAB1, SELP, LOC107987479

-
22q11.2 Deletion Syndrome
Medlineplus
The features of this syndrome vary widely, even among affected members of the same family. ... Doctors named these conditions DiGeorge syndrome, velocardiofacial syndrome (also called Shprintzen syndrome), and conotruncal anomaly face syndrome. In addition, some children with the 22q11.2 deletion were diagnosed with the autosomal dominant form of Opitz G/BBB syndrome and Cayler cardiofacial syndrome. ... Frequency 22q11.2 deletion syndrome affects an estimated 1 in 4,000 people. ... The loss of additional genes in the deleted region likely contributes to the varied features of 22q11.2 deletion syndrome. Learn more about the genes and chromosome associated with 22q11.2 deletion syndrome COMT TBX1 chromosome 22 Inheritance Pattern The inheritance of 22q11.2 deletion syndrome is considered autosomal dominant because a deletion in one copy of chromosome 22 in each cell is sufficient to cause the condition.TBX1, COMT, CRKL, FGF8, HIRA, DGCR, DGCR6, DGCR2, DGCR8, UFD1, GP1BB, JMJD1C, RREB1, ARVCF, ESS2, SEC24C, FOXN1, HOXA3, TGFBR2, DOCK1, DICER1, VEGFA, PLXND1, NDST1, ALDH1A2, KAT6A, CHRD, ZNF366, CHD7, PRODH, DGS2, HTC2, GSC2, TBX5, FBXO7, HTRA2, SLC25A1, ZNF74, CDC45, SEPTIN5, GNB1L, LRRK2, CECR7, SCZD12, CELF2, MIR185, MIR132, BCRP2, FOXI3, CIC, DNAJC13, GCOM1, SPECC1L, NKX2-6, CPO, RAB39B, MED15, DERL3, DGCR6L, SHANK3, SYVN1, DGCR5, C19orf12, TCL6, POLR2M, PINK1, NAAA, PRDM9, NPLOC4, CECR2, AFP, ZAP70, APOL1, CTNNB1, IGLC2, IGLC1, GBA, GATA3, EYA1, ELN, EDN1, MAP3K8, IGLC4, CLTC, CHRNA7, CD27, BMP4, BCR, BCL2, ARSA, IGLC3, IGLC5, PLA2G6, SULT1E1, LZTR1, DVL1P1, CDR3, ALB, TNF, TJP1, TBX3, SNCA, IGLC6, SLC7A4, SIX1, ATXN2, RANBP1, RAC2, LAMC1, IL2, MYZAP

-
Mirror Syndrome
Wikipedia
Mirror syndrome or triple edema or Ballantyne syndrome is a rare disorder affecting pregnant women. ... Platelet count , aspartate transaminase , alanine transaminase , and haptoglobin are usually unaffected and may be used to distinguish mirror syndrome from HELLP syndrome . [6] [10] [11] [12] [13] Treatment [ edit ] In most cases Ballantyne syndrome causes fetal or neonatal death and, in contrast, maternal involvement is limited at the most to preeclampsia . ... "Clinical biological features of Ballantyne syndrome and the role of placental hydrops". ... "Selective fetocide reversed mirror syndrome in a dichorionic triplet pregnancy with severe twin-twin transfusion syndrome: a case report". ... "Spontaneous reversal of mirror syndrome in a twin pregnancy after a single fetal death".
-
Clinodactyly
Wikipedia
It is a fairly common isolated anomaly which often goes unnoticed, but also occurs in combination with other abnormalities in certain genetic syndromes . [1] The term is from the Ancient Greek κλίνειν klínein meaning "to bend" and δάκτυλος dáktulos meaning "digit". Contents 1 Genetics 2 Pathophysiology 3 Diagnosis 4 Management 5 Epidemiology 6 See also 7 References 8 External links Genetics [ edit ] Clinodactyly is an autosomal dominant trait that has variable expressiveness and incomplete penetrance . [ citation needed ] Clinodactyly can be passed through inheritance and presents as either an isolated anomaly or a component manifestation of a genetic syndrome. [2] Many syndromes are associated with clinodactyly, including those listed below, [ citation needed ] , but the phenotype, by itself, is not a sensitive or specific diagnostic test for these syndromes (it is present in up to 18% of the normal population [3] ). Down syndrome Turner syndrome Aarskog syndrome Carpenter syndrome Seckel syndrome Cornelia de Lange syndrome Orofaciodigital syndrome 1 13q deletion syndrome XXYY syndrome Silver–Russell syndrome Andersen-Tawil syndrome Noonan syndrome Ehlers–Danlos syndrome When identified prenatally, in conjunction with other features of Down syndrome, for example during obstetric ultrasonography , it may be an indication for intrauterine sampling for fetal chromosome analysis. [3] Pathophysiology [ edit ] Due to a developmental arrest, there is an abnormal alignment of the joint surfaces at either interphalangeal joint causing angulation in the plane of the palm. ... Reports of incidence vary between 1% and 19.5%. [1] See also [ edit ] Camptodactyly OSLAM syndrome References [ edit ] ^ a b c d Flatt, Adrian E (October 2005). ... External links [ edit ] Classification D ICD - 10 : Q74.0 ICD - 9-CM : 755.59 DiseasesDB : 16756 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatumTFAP2B, ABL1, TFAP2A, TBX22, PHGDH, SIN3A, NSMF, TCTN3, KIFBP, INTU, NCAPG2, PGAP2, CCDC22, ANKRD11, NIN, POMP, MAGEL2, POC1A, NPAP1, CLCF1, IRX5, MED12, HUWE1, MKRN3-AS1, ALG3, CD96, SF3B4, SLC35A1, MED13L, COLEC10, STAMBP, MAN1B1, PUF60, CHSY1, SPART, BCOR, MKS1, ALX3, PHIP, SPRTN, KIAA1109, TRAF7, PIGO, PIGY, G6PC3, PGAP3, PWAR1, ESCO2, ARID2, PIGW, SNORD115-1, HEPHL1, KIF7, PWRN1, ITCH, FBXO11, FAT4, NUP107, SLC29A3, PIGV, IFT122, ERMARD, KLHL7, KIF15, SNX14, BCL2L14, WDR35, TRPV4, PIEZO2, XYLT1, NXN, CPLANE1, CEP57, PUM1, PIGL, IPW, GJA1, GJB2, IGF1, IGF1R, INS, PDX1, KCNJ11, PIK3R1, LMNA, LRP6, SMAD4, NDN, ROR2, TRIP12, GCK, FGD1, GPC4, BPTF, EZH2, DVL3, DVL1, DDX11, CSNK2A1, COL2A1, CDH1, CDC42, CDC6, BRAF, BRCA1, ATRX, ATR, PAX3, PAH, POLE, WNT5A, WNT7A, MKRN3, PCGF2, RBM10, KDM5C, SMC1A, NAA10, OFD1, CDK10, CDK13, DCHS1, CACNA1G, HERC2, RAB11B, NOG, POR, SNORD116-1, TRIO, SOX11, MAP2K1, PTPN11, DPF2, RPL10, RPS6KA3, SNRPN, SOX4, SOX5, SSR4, ABCC8, TCF4, TNNT3, TGDS, KCNJ2, WDR20, IGF2, PHF6, FGFR2