Critical Care Nursing Clinics of North America . 28 (3): 309–16. doi : 10.1016/j.cnc.2016.04.005 . ... Archived from the original on 2008-12-16 . Retrieved 2008-11-28 . ^ Mathew PK (January 1981). "Diving reflex.
Exchange transfusions were required for fetal anemia at 28 and 30 weeks' gestation. Transfusions were administered at birth by cesarean section at week 35 and at regular intervals thereafter. ... All but 1 of the mutations were located in exons 12 to 28; 1 mutation was found in exon 6. Population Genetics The R1042W mutation in the CDAN1 gene (607465.0001) is a founder mutation in the Bedouin population (Tamary et al., 2008).
In 60% of affected individuals two pathogenic variants were identified by sequence analysis, in 28% only one pathogenic variant was identified, and in 11% no pathogenic variant was identified (Note: Testing to detect splice site variants and large deletions was not performed) [Authors and other labs, combined data, unpublished]. 8.
A number sign (#) is used with this entry because congenital dyserythropoietic anemia type Ib (CDAN1B) is caused by homozygous mutation in the C15ORF41 gene (615626) on chromosome 15q14. Description Congenital dyserythropoietic anemia type I is an autosomal recessive hematologic disorder characterized by congenital macrocytic anemia secondary to ineffective erythropoiesis. The bone marrow shows erythroid hyperplasia, with nuclear abnormalities in most erythroblasts. Up to 3% of erythroblasts have interchromatin bridges, and erythroblast nuclei are abnormally electron dense with spongy ('Swiss cheese-like') heterochromatin on electron microscopy. Some reported patients have distal digital abnormalities (summary by Ahmed et al., 2006).
Congenital dyserythropoietic anemia (CDA) type 1 is an inherited blood disorder characterized by moderate to severe anemia . It is usually diagnosed in childhood or adolescence, although in some cases, the condition can be detected before birth. Many affected individuals have yellowing of the skin and eyes (jaundice) and an enlarged liver and spleen (hepatosplenomegaly). This condition also causes the body to absorb too much iron, which builds up and can damage tissues and organs. In particular, iron overload can lead to an abnormal heart rhythm (arrhythmia), congestive heart failure, diabetes, and chronic liver disease (cirrhosis).
Congenital dyserythropoietic anemia (CDA) is an inherited blood disorder that affects the development of red blood cells. This disorder is one of many types of anemia , which is a condition characterized by a shortage of red blood cells. This shortage prevents the blood from carrying an adequate supply of oxygen to the body's tissues. The resulting symptoms can include tiredness (fatigue), weakness, pale skin, and other complications. Researchers have identified three major types of CDA: type I, type II, and type III.
Congenital dyserythropoietic anemiatype I (CDA I) is a hematologic disorder of erythropoiesis characterized by moderate to severe macrocytic anemia occasionally associated with limb or nail deformities and scoliosis. Epidemiology The prevalence is unknown. Over a 42 year period (from 1967-2009), 122 CDA I cases were reported in Europe. Clinical description CDA I usually presents in the first decade of life. Manifestations include a moderate macrocytic anemia associated with intermittent jaundice, frequent splenomegaly and occasional hepatomegaly. Approximately 1/3 of CDA I patients may also have congenital malformations of the limbs (supernummary toes, hand or feet syndactyly, absence of nails) or heart (ventricular septal defect), double kidneys, short stature or hip dysplasia.
Congenital dyserythropoietic anemia type I (CDA I) is a disorder of blood cell production, particularly of the production of erythroblasts, which are the precursors of the red blood cells (RBCs). [1] Congenital dyserythropoietic anemia type I Specialty Hematology Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Treatment 5 See also 6 References 7 Further reading 8 External links Presentation [ edit ] Many affected individuals have yellowing of the skin and eyes (jaundice) and an enlarged liver and spleen (hepatosplenomegaly). This condition also causes the body to absorb too much iron, which builds up and can damage tissues and organs. In particular, iron overload can lead to an abnormal heart rhythm (arrhythmia), congestive heart failure, diabetes, and chronic liver disease (cirrhosis). Rarely, people with CDA type I are born with skeletal abnormalities, most often involving the fingers and/or toes. [2] Genetics [ edit ] CDA type I is transmitted by both parents autosomal recessively and usually results from mutations in the CDAN1 gene. Little is known about the function of this gene, and it is unclear how mutations cause the characteristic features of CDA type I.
. ^ Prasad, Kameshwar; Singh, Mamta B; Ryan, Hannah (28 April 2016). "Corticosteroids for managing tuberculous meningitis" . ... ISBN 9780199234073 . ^ Central Nervous System Bacterial Infections—Advances in Research and Treatment: 2012 Edition: ScholarlyBrief . ScholarlyEditions. 2012-12-26. p. 28. ISBN 9781481616232 . ^ Rajshekhar, Vedantam (2009).
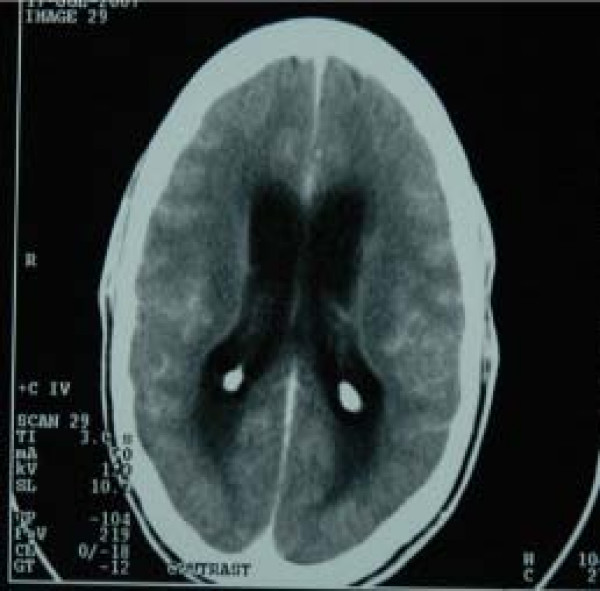
A rare bacterial infectious disease caused by Mycobacterium tuberculosis , characterized by a variable clinical picture comprising classic manifestations of meningitis, i. e. headache, fever, and stiff neck, in addition to cranial nerve palsies (most commonly III, VI, and VII), altered mental status, and seizures, among others. Basal meningeal enhancement in neuroimaging, cerebrospinal fluid abnormalities (moderate lymphocytic pleocytosis, moderately elevated protein concentration, low glucose), and a chest x-ray suggestive of pulmonary tuberculosis may support the diagnosis.
The New England Journal of Medicine . 358 (12): 1215–28. doi : 10.1056/NEJMoa070812 . PMID 18344568 . ... "Treatment of patients with the hypereosinophilic syndrome with mepolizumab" . N. Engl. J. Med . 358 (12): 1215–28. doi : 10.1056/NEJMoa070812 . PMID 18344568 . ^ Cools J, DeAngelo DJ, Gotlib J, et al. (2003).
Hypereosinophilic syndrome (HES) refers to a rare group of conditions that are associated with persistent eosinophilia with evidence of organ involvement. Signs and symptoms vary significantly based on which parts of the body are affected. Although any organ system can be involved in HES, the heart, central nervous system, skin, and respiratory tract are the most commonly affected. The condition was originally thought to be "idiopathic" or of unknown cause. However, recent advances in diagnostic testing have allowed a cause to be identified in approximately a quarter of cases.
Overview Hypereosinophilic (hy-per-ee-o-SIN-o-phil-ik) syndrome (HES) is a group of blood disorders that occur when you have high numbers of eosinophils — white blood cells that play an important role in your immune system. Over time, the excess eosinophils enter various tissues, eventually damaging your organs. The most common targets are the skin, lungs, digestive tract, heart, blood and nervous system. Untreated, HES can become life-threatening. Symptoms Early symptoms of HES may include fatigue, cough, breathlessness, muscle pain, rash and fever. Causes Some varieties of hypereosinophilic syndrome tend to run in families.
Hypereosinophilic syndrome (HES) constitutes a rare and heterogeneous group of disorders, defined as persistent and marked blood eosinophilia and/or tissue eosinophilia associated with a wide range of clinical manifestations reflecting eosinophil-induced tissue/organ damage. Epidemiology Prevalence is unknown. It frequently occurs in middle-aged patients, but may concern any age group. Clinical description Target-organ damage mediated by eosinophils is highly variable among patients, and consists of dermatological involvement (urticaria, eczema, angioedema, pruriginous papules, nodules, erythroderma) in more than 50% of cases, followed by involvement of lungs (cough, breathlessness and wheezing) and digestive tract (nausea, vomiting, abdominal pain, diarrhea, ascites) in roughly 40%. Cardiac involvement is less frequent, but must be recognized early due to irreversible and life-threatening complications such as acute myocarditis, intraventricular thrombus, endomyocardial fibrosis and valve thickening and/or destruction. Constitutional symptoms of fever, myalgia and fatigue may occur. Other common complications include central or peripheral nervous system involvement, hepato- and/or splenomegaly, and coagulation disorders.
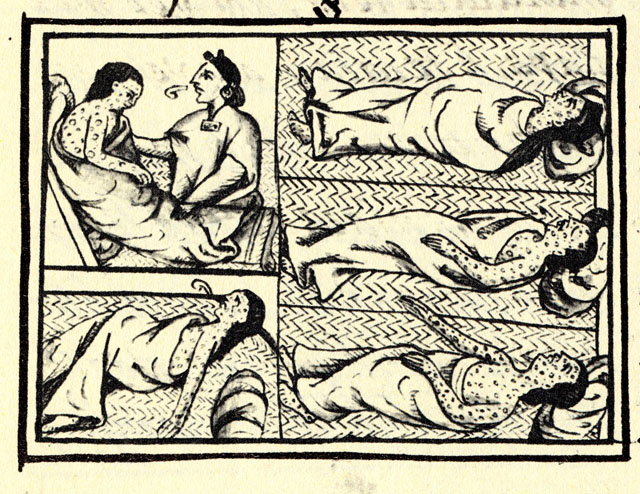
Paratyphi C continues to cause enteric fevers, and if untreated, has a mortality rate up to 15%. [28] Infections are largely limited to developing nations in Africa and Asia, although enteric fevers, in general, are still a health threat world wide. [29] Infections with S. ... Paratyphi C may have first transferred over to humans from swine in the Old World during, or, shortly after the Neolithic period. [30] Some, including evolutionary geneticist, María Ávila-Arcos, have questioned this evidence, since S. enterica symptoms are poorly matched with the disease. [31] [32] [28] Both Ávila-Arcos, and even Krause’s team and authors of earlier historical analyses, [33] point out that RNA viruses , among other non-bacterial pathogens, had not been investigated. ... Latin American Antiquity . 23 (1): 3–28. doi : 10.7183/1045-6635.23.1.3 . ^ a b c d e Marr, John; Kiracofe, J.B. (2000). ... External links [ edit ] "Megadeath in Mexico" . discovermagazine.com . Retrieved 2015-11-28 . " ' Ebola' bug wiped out the Aztecs" . The Guardian . Retrieved 2015-11-28 . "Expert: Native disease killed Aztecs, not outsiders" . chron.com .
Moran states that it would be unwise routinely to dismiss anti-E as being of little clinical consequence. [2] In the case of anti-E, the woman should be checked around 28 weeks to see if she has developed anti-c as well. [ citation needed ] Testing [ edit ] Testing for HDN involves blood work from both mother and father, and may also include assessment with amniocentesis and Middle Cerebral Artery scans. ... After that, the risk of an IUT is greater than the risk from post birth transfusion. [28] Steroids – steroids are sometimes given to the mother before IUTs and early delivery to mature the fetal lungs. [28] [15] Phenobarbital – Phenobarbital is sometimes given to the mother to help mature the fetal liver and reduce hyperbilirubinemia. [15] [29] Early delivery – delivery can occur anytime after the age of viability. [1] Emergency delivery due to failed IUT is possible, along with induction of labor at 35–38 weeks. [28] [30] After birth [ edit ] Testing [ edit ] Coombs – after birth the baby will have a direct Coombs test run to confirm antibodies attached to the infant's red blood cells. ... Ultrasound in Obstetrics and Gynecology . 28 (6): 814–20. doi : 10.1002/uog.2837 .
Some argue that humans evolved to avoid ketosis and should not be in ketosis long-term. [26] The counter-argument is that there is no physiologic requirement for dietary carbohydrate as adequate energy can be made via gluconeogenesis and ketogenesis indefinitely. [27] Alternatively, the switching between a ketotic and fed state has been proposed to have beneficial effects on metabolic and neurologic health. [4] The effects of sustaining ketosis for up to two years are known from studies of people following a strict ketogenic diet for epilepsy or type 2 diabetes, and these including short-term adverse effects leading to potential long-term ones. [28] However, literature on longer term effects or intermittent ketosis is lacking. [28] Medication considerations [ edit ] Some medications require attention when in a state of ketosis, especially several classes of diabetes medication. ... This usually occurs with missed insulin doses, illness, dehydration or adherence to a low-carbohydrate diet while taking the medication. [29] Additionally, medications used to directly lower blood glucose including insulin and sulfonylureas may cause hypoglycemia if they are not titrated prior to starting a diet that results in ketosis. [28] Adverse effects [ edit ] The most common side effects of ketosis include headache, fatigue, dizziness, insomnia, difficulty in exercise tolerance, constipation, and nausea, especially in the first days and weeks after starting a ketogenic diet. [28] Breath may develop a sweet, fruity flavor via production of acetone that is exhaled because of its high volatility. [10] Most adverse effects of long-term ketosis reported are in children because of its longstanding acceptance as a treatment for pediatric epilepsy. ... Journal of Lipid Research . 55 (11): 2211–28. doi : 10.1194/jlr.R048975 . PMC 4617125 .
Choreoathetosis is also a common presentation of dyskinesia as a side effect of levodopa-carbidopa in the treatment of Parkinson disease. [1] The use of crack cocaine can result in a condition of crack dancing described as choreathetoid. [2] See also [ edit ] Ulegyria References [ edit ] ^ "28". Basic and Clinical Pharmacology (Eleventh ed.).
Proband 1333, who had 4 miscarriages followed by 4 hydatidiform moles, and her 2 sisters, who had 1 and 3 miscarriages, respectively, were homozygous for a nonsense mutation in exon 28 (W1151X; 608797.0001). Proband 880, with 6 miscarriages and 1 complete hydatidiform mole, and her brother, with nonobstructive azoospermia and no Y chromosome deletions, were compound heterozygous for an invariant splice site mutation (608797.0002) and a 1-bp deletion (608797.0003).
The Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators (2016) examined sequence data for the 7 protein-coding exons of ANGPTL4 in 6,924 patients with early-onset myocardial infarction and 6,834 controls, and detected 10 variants predicted to result in loss of gene function, carried by 28 heterozygous individuals. Carriers of loss-of-function alleles had significantly lower levels of triglycerides than did noncarriers (mean, 35% lower; p = 0.003), with no significant differences in LDL or HDL cholesterol levels.
The deletion was not found in either parent, who had normal echocardiograms at ages 28 and 34 years, respectively. The girl also carried a known MYBPC3 (600958) polymorphism, V896M, which was also found in her unaffected father; the authors suggested that the V896M variant may have acted as a modifier, exacerbating the phenotypic expression of the TNNT2 mutation and causing an unusually early onset of RMC.
Wang et al. (2015) reported a multigenerational Chinese family with onset of sensorineural hearing loss between 10 and 28 years of age. Only 1 patient had onset at age 3, and this was associated with aminoglycoside exposure.
Treatment with levodopa was not beneficial. Fuchs et al. (2013) reported 28 patients from 8 families with autosomal dominant primary torsion dystonia, including the family reported by Bressman et al. (1994), which was referred to as family D1.
Heterozygous DYT-GNAL Age of onset. In the 28 individuals first described by Fuchs et al [2013], mean age at disease onset was 31.3 years (± 12.4 years); range: 7-54 years. ... Generalized dystonia is far less common. In a study of 28 individuals, dystonia remained focal in 12 and became segmental in 13 or generalized in three [Fuchs et al 2013]. ... Speech involvement was reported in 44% of 28 patients [Fuchs et al 2013]. Dystonic tremor.
Blood pressure decreased early in the first pregnancy of 1 carrier, but then rose dramatically, reaching 170/130 mm Hg at 28 weeks despite antihypertensive therapy.
Hypertension due to gain-of-function mutations in the mineralocorticoid receptor is a rare genetic hypertension characterized by a familial severe hypertension with an onset before age 20 years, associated with suppressed plasma renin and low aldosterone levels in the presence of low or normal levels of the mineralocorticoid aldosterone, that is highly resistant to antihypertensive medication. During pregnancy, there is a marked exacerbation of hypertension, accompanied by low serum potassium levels and undetectable aldosterone levels, but without signs of preeclampsia, requiring early delivery.
The mutation was found in 11 (13%) of 84 patients who underwent exome sequencing and in 2 (7%) of 28 patients who underwent direct sequencing of the KCNC1 gene.
A rare, genetic, neurological disorder characterized by childhood to adolescent onset of progressive myoclonus (which becomes very severe and results in major motor impediment) associated with infrequent tonic-clonic seizures, and, occasionally, ataxia. Learning disability prior to seizure onset and mild cognitive decline may be associated.
Parkinson's Disease: A Guide to Patient Care . Springer Publishing Company. p. 28. ISBN 978-0-8261-2269-8 . Bibliography [ edit ] O'Sullivan, Susan; Schmitz, Thomas (2007).
Compendium on Continuing Education for the Practicing Veterinarian . Veterinary Learning Systems. 28 (5): 373–382. ^ Ouellet M, Dunn M, Lussier B, Chailleux N, Hélie P (2006).
"Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases". Am J Surg Pathol . 16 (9): 815–37. doi : 10.1097/00000478-199209000-00001 .
A very rare, malignant, epithelial tumor of the pancreas characterized, macroscopically, by a usually large, well-circumscribed, fully or partially encapsulated, solid mass, often with hemorrhage, necrosis and cystic changes, in any portion of the pancreas and, histologically, by neoplastic cells with variable degrees of differentiation and morphology, ranging from acinar structures similar to normal pancreatic acini to large sheets of poorly differentiated neoplastic cells. Presenting symptoms are typically non-specific and include abdominal pain, weight loss, vomiting, nausea, and/or, less commonly, jaundice. Immunohistochemical evidence of acinar-specific products is observed. Association with Lynch syndrome, familial adenomatous polyposis, and pancreatic panniculitis has been reported.
A rare genetic eye disease characterized by optic disc anomalies (bilateral colobomatous optic discs, retinal vessels arising from the peripheral optic disc) and macular atrophy. Peripapillary chorioretinal atrophy and chorioretinal and iris coloboma have also been described. Patients present with horizontal nystagmus and poor visual acuity.