It shares phenotypic features with Joubert syndrome (see 213300), COACH syndrome (216360), and familial juvenile nephronophthisis (see 256100). ... Matsuzaka et al. (1986) concluded that the constellation of findings was consistent with a distinct clinicopathologic entity, which they termed 'cerebro-oculo-hepato-renal syndrome,' or 'Arima syndrome.' Lindhout et al. (1980) reported a case of Joubert syndrome with associated bilateral chorioretinal coloboma, which the authors noted had not previously been reported in Joubert syndrome. ... The conclusions were based on 72 previously reported cases and 29 new cases with the possible diagnosis of Joubert syndrome. Saraiva and Baraitser (1992) suggested that the form of Joubert syndrome with retinal dystrophy could be called Dekaban syndrome, since it was first reported by Dekaban (1969). ... Lewis et al. (1994) suggested that Joubert syndrome is a member of a spectrum of congenital malformation syndromes involving the CNS, eye, liver, and kidneys. ... They emphasized the phenotypic overlap and diagnostic difficulties among Arima syndrome, Joubert syndrome, COACH syndrome, and some forms of familial nephronophthisis.
A number sign (#) is used with this entry because Joubert syndrome-14 (JBTS14) is caused by homozygous or compound heterozygous mutation in the TMEM237 gene (614423) on chromosome 2q33. ... For a phenotypic description and a discussion of genetic heterogeneity of Joubert syndrome, see 213300. Clinical Features Boycott et al. (2007) reported 10 patients with Joubert syndrome in a Canadian Hutterite population. ... Seven of the patients had previously been diagnosed as having Meckel syndrome (see, e.g., 249000). Boycott et al. (2007) also reviewed a report of 3 Hutterite patients reported as having Meckel syndrome (Schurig et al., 1980) and concluded that all had a variant of Joubert syndrome. Mutations and deletions in the NPHP1 gene, as well as evaluation of known loci for Meckel and Joubert syndrome, were all excluded, suggesting further genetic heterogeneity. ... Inheritance The transmission pattern of Joubert syndrome-14 in the families reported by Huang et al. (2011) was consistent with autosomal recessive inheritance.
A rare subtype of Joubert syndrome (JS) and related disorders (JSRD) characterized by the neurological features of JS associated with both renal and ocular disease.
Heterozygous mutation in the ZNF423 gene causes Joubert syndrome-19 (JBTS19). For a general phenotypic description and a discussion of genetic heterogeneity of nephronophthisis, see NPHP1 (256100). For a phenotypic description and a discussion of genetic heterogeneity of Joubert syndrome, see JBTS1 (213300). Clinical Features Chaki et al. (2012) reported 2 Turkish sibs, born of consanguineous parents (family F824), with infantile-onset NPHP, cerebellar vermis hypoplasia, and situs inversus. ... Chaki et al. (2012) also reported 2 unrelated patients with Joubert syndrome. One patient had cerebellar vermis hypoplasia, Leber congenital amaurosis (see 204000), and polycystic kidney disease. ... Inheritance The transmission pattern of NPHP14 in the family reported by Chaki et al. (2012) was consistent with autosomal recessive inheritance. Two patients with Joubert syndrome-19 were found to carry heterozygous ZNF423 mutations, suggesting autosomal dominant inheritance. ... Two of 96 additional patients with Joubert syndrome were found to carry heterozygous ZNF423 mutations (604557.0002 and 604557.0003).
A number sign (#) is used with this entry because of evidence that Joubert syndrome-9 (JBTS9) is caused by homozygous or compound heterozygous mutation in the CC2D2A gene (612013) on chromosome 4p15. Digenic inheritance has also been reported; see MOLECULAR GENETICS. Meckel syndrome-6 (MKS6; 612284) is an allelic disorder. ... Gorden et al. (2008) noted that the features in this patient were reminiscent of COACH syndrome (216360), which in some cases may be a variant representing a transitional phenotype between Joubert syndrome and Meckel syndrome (249000). ... Patients with CC2D2A-related Joubert syndrome were more likely to have ventriculomegaly and seizures compared to Joubert syndrome patients without CC2D2A mutations. Although mutation-specific genotype-phenotype correlations could not be identified, most Joubert syndrome patients had at least 1 missense mutation, compared to those with the more severe Meckel syndrome, which tends to be associated with nonsense mutations (see 612013.0002).
A number sign (#) is used with this entry because of evidence that Joubert syndrome-5 (JBTS5) is caused by homozygous or compound heterozygous mutation in the gene encoding the centrosomal protein CEP290 (610142) on chromosome 12q21. For a phenotypic description and a discussion of genetic heterogeneity of Joubert syndrome, see 213300. Clinical Features Joubert syndrome (JBTS) is an autosomal recessive disorder presenting with psychomotor delay, hypotonia, ataxia, oculomotor apraxia, and neonatal breathing abnormalities (Valente et al., 2006). ... A number of distinct syndromes sharing the MTS have been described, presenting wide phenotypic variability both within and among families (Gleeson et al., 2004). Valente et al. (2006) described the JBTS5 phenotype as characterized mainly by the neurologic and neuroradiologic features of Joubert syndrome associated with severe retinal and renal involvement, but noted that the clinical spectrum was broad, including incomplete phenotypes such as cerebelloretinal and cerebellorenal syndromes. ... Valente et al. (2006) identified the JBTS5 locus on 12q21.31-q21.33 in linkage analysis from consanguineous families with Joubert syndrome-related disorders (JSRDs), which showed the neurologic features of Joubert syndrome associated with multiorgan involvement (mainly retinal dystrophy and nephronophthisis).
Mutation in the TMEM216 gene can also cause Meckel syndrome-2 (MKS2; 603194). For a phenotypic description and a discussion of genetic heterogeneity of Joubert syndrome, see 213300. ... In affected members of 14 families with Joubert syndrome-2 and 6 families with Meckel syndrome-2 (MKS2; 603194), Valente et al. (2010) identified 7 different homozygous mutations in the TMEM216 gene (see, e.g., 613277.0001-613277.0004). ... A Turkish family had 2 affected patients: 1 with Joubert syndrome and a fetus with Meckel syndrome, indicating that the 2 clinical disorders are part of the same spectrum. Nomenclature Keeler et al. (2003) noted that the form of Joubert syndrome that includes retinal dysplasia and cystic dysplastic kidneys has been differentiated from other forms of Joubert syndrome and called either Joubert syndrome type B or type 2. ... Likewise, Valente et al. (2003) proposed the CORS acronym for loci identified in all cerebellooculorenal syndromes sharing the pathognomonic MTI, including Joubert syndrome and incomplete phenotypes such as COGAN (257550) and cerebellorenal syndromes.
A number sign (#) is used with this entry because Joubert syndrome-16 (JBTS16) can be caused by homozygous mutation in the TMEM138 gene (614459) on chromosome 11q12. Description Joubert syndrome-16 is an autosomal recessive developmental disorder characterized by the molar tooth sign on brain imaging, oculomotor apraxia, variable coloboma, and rare kidney involvement. ... For a phenotypic description and a discussion of genetic heterogeneity of Joubert syndrome, see 213300. Clinical Features Lee et al. (2012) reported 8 consanguineous Arab families with Joubert syndrome. ... Inheritance The transmission pattern of Joubert syndrome in the families reported by Lee et al. (2012) was consistent with autosomal recessive inheritance. Molecular Genetics By repeat sequencing of candidate genes in 6 consanguineous Arab families with Joubert syndrome showing linkage to the JBTS2 locus (608091) on chromosome 11q13, but who were negative for mutations in the TMEM216 gene (613277), Lee et al. (2012) identified homozygous mutations in the TMEM138 gene (614459.0001-614459.0005).
The various forms of Ehlers-Danlos syndrome have been classified in several different ways. ... The 2017 classification describes 13 types of Ehlers-Danlos syndrome. An unusually large range of joint movement (hypermobility) occurs in most forms of Ehlers-Danlos syndrome, and it is a hallmark feature of the hypermobile type. ... During pregnancy, women with vascular Ehlers-Danlos syndrome may experience rupture of the uterus. ... Other types of Ehlers-Danlos syndrome have additional signs and symptoms. ... Other rare forms of Ehlers-Danlos syndrome result from mutations in other genes.
A number sign (#) is used with this entry because of evidence that brittle cornea syndrome-2 (BCS2) is caused by homozygous mutation in the PRDM5 gene (614161) on chromosome 4q27. Description Brittle cornea syndrome (BCS) is characterized by blue sclerae, corneal rupture after minor trauma, keratoconus or keratoglobus, hyperelasticity of the skin, and hypermobility of the joints (Al-Hussain et al., 2004). It is classified as a form of Ehlers-Danlos syndrome (Malfait et al., 2017). For a discussion of genetic heterogeneity of brittle cornea syndrome, see BCS1 (229200). ... Molecular Genetics In affected members of a consanguineous Pakistani family (BC-001) with brittle cornea syndrome mapping to chromosome 4, Burkitt Wright et al. (2011) identified a 52.46-kb homozygous deletion involving exons 9 to 14 of the PRDM5 gene (614161.0001). ... Porter et al. (2015) identified homozygosity for PRDM5 mutations in 2 additional patients with brittle cornea syndrome, and restudied patients from the Pakistani families previously reported by Burkitt Wright et al. (2011).
A number sign (#) is used with this entry because brittle cornea syndrome-1 (BCS1) is caused by homozygous mutation in the ZNF469 gene (612078) on chromosome 16q24. ... It is classified as a form of Ehlers-Danlos syndrome (Malfait et al., 2017). Genetic Heterogeneity of Brittle Cornea Syndrome Brittle cornea syndrome-2 (BCS2; 614170) is caused by mutation in the PRDM5 gene (614161) on chromosome 4q27. ... The affected sibs did not present any signs of Marfan syndrome (see 154700) or Ehlers-Danlos syndrome, and there was no family history of bone fragility. ... Al-Hussain et al. (2004) described 23 patients with brittle cornea syndrome from 13 Middle Eastern families (9 from Saudi Arabia, 2 from Syria, 1 from Jordan, and 1 from Yemen). ... Khan et al. (2010) reported a consanguineous Syrian family in which 2 sibs had brittle cornea syndrome and 1 sib had blue sclerae only.
A rare, hereditary connective tissue disease characterized by severe ocular manifestations due to extreme corneal thinning and fragility with rupture in the absence of significant trauma, often leading to irreversible blindness. Extraocular manifestations comprise deafness, developmental hip dysplasia, and joint hypermobility.
There are two forms of Opitz G/BBB syndrome, X-linked Opitz G/BBB syndrome and autosomal dominant Opitz G/BBB syndrome. ... The incidence of autosomal dominant Opitz G/BBB syndrome is unknown. It is part of a larger condition known as 22q11.2 deletion syndrome, which is estimated to affect 1 in 4,000 people. Causes X-linked Opitz G/BBB syndrome is caused by mutations in the MID1 gene. ... Because this same region is deleted in another condition called 22q11.2 deletion syndrome, researchers often consider Opitz/GBBB syndrome caused by this genetic change to be a form of 22q11.2 deletion syndrome. It is not known which of the deleted genes contribute to the signs and symptoms of Opitz G/BBB syndrome. In other people, autosomal dominant Opitz/GBBB syndrome is caused by a mutation in the SPECC1L gene, which is near the 22q11.2 region but is not in the area that is typically deleted in other individuals with autosomal dominant Opitz G/BBB syndrome or 22q11.2 deletion syndrome.
Penetrance Usually the presence of an MID1 pathogenic variant is associated with clinical findings of X-OS; however, recently an instance of reduced penetrance has been reported [Ruiter et al 2010]. Nomenclature Opitz G/BBB syndrome was first reported as two separate entities, BBB syndrome [Opitz et al 1969b] and G syndrome [Opitz et al 1969a]. Subsequently, it has become apparent that the two syndromes identified in 1969 are in fact a single entity, now named Opitz G/BBB syndrome. Other names, no longer used, include hypospadias-dysphagia syndrome, Opitz-Frias syndrome, telecanthus with associated abnormalities, and hypertelorism-hypospadias syndrome. Of note, X-linked Opitz G/BBB syndrome (X-OS; OSX; type I) is distinct from autosomal dominant Opitz G/BBB syndrome (ADOS; type II). ... Many males with X-linked Opitz G/BBB syndrome require special educational programs.
Description The Opitz GBBB syndrome is a congenital midline malformation syndrome characterized by hypertelorism, hypospadias, cleft lip/palate, laryngotracheoesophageal abnormalities, imperforate anus, developmental delay, and cardiac defects (So et al., 2005). This disorder was first reported as 2 separate entities, BBB syndrome and G syndrome; subsequent reports of families in which the BBB and G syndromes segregated within a single kindred suggested that they represent a single entity. ... Neither of these defects had previously been reported in association with Opitz syndrome. Since both were midline defects, they further characterized Opitz syndrome as an impairment in midline development. ... Trockenbacher et al. (2001) pointed out that the IGBP1 gene maps to the same linkage interval in Xq13 as does the FG syndrome (305450). Similar to Opitz syndrome, FG syndrome is characterized by mental retardation combined with imperforate anus, congenital heart defects, and characteristic facies. ... So et al. (2005) demonstrated the wide spectrum of severity and manifestations of Opitz syndrome, and they emphasized that X-linked Opitz syndrome patients with MID1 mutations may be less severely affected than previously reported.
Opitz G/BBB syndrome is an inherited condition that affects several structures along the midline of the body. ... There are two forms of Opitz G/BBB syndrome, which are distinguished by their genetic causes and patterns of inheritance. The X-linked form is caused by mutations in the MID1 gene. Autosomal dominant Opitz G/BBB syndrome is caused by a deletion of 22q11.2, and is often referred to as 22q11.2 deletion syndrome .
Deletion in the same region may also result in DiGeorge syndrome (188400) and velocardiofacial syndrome (192430). ... The Opitz GBBB syndrome was earlier thought to be 2 separate X-linked syndromes called the G syndrome and the BBB syndrome; both were listed in the X-linked catalog as recently as the seventh edition of MIM (1986). ... Nomenclature Once it was known that the G syndrome and the BBB syndrome were in fact the same, Sedano and Gorlin (1988) suggested that the condition be called the Opitz oculogenitolaryngeal syndrome. ... Parisian and Toomey (1978) suggested that the G syndrome and the BBB syndrome are identical. ... Based on a comparison of Opitz-GBBB syndrome with the chromosome 22 microdeletion syndrome, McDonald-McGinn et al. (1995) suggested that autosomal dominant Opitz-GBBB syndrome may be the result of a 22q11.2 deletion in some cases.
Hyper IgM syndrome type 4 Immunoglobulin M Specialty Hematology Types Hyper-IgM syndrome type 1,2,3,4 and 5 [1] [2] [3] [4] [5] Diagnostic method MRI, Chest radiography and genetic testing [6] Treatment Allogeneic hematopoietic cell transplantation [7] Hyper-IgM syndrome type 4 is a form of Hyper IgM syndrome which is a defect in class switch recombination downstream of the AICDA gene that does not impair somatic hypermutation . [8] Contents 1 Hyper IgM syndromes 2 Signs and symptoms 3 Cause 4 Pathophysiology 5 Diagnosis 6 Treatment 7 References Hyper IgM syndromes [ edit ] Hyper IgM syndromes is a group of primary immune deficiency disorders characterized by defective CD40 signaling; via B cells affecting class switch recombination (CSR) and somatic hypermutation . ... As a consequence, people with HIGM have an increased susceptibility to infections. [9] [7] [10] Signs and symptoms [ edit ] Hyper IgM syndrome can have the following syndromes: [6] [11] Infection / Pneumocystis pneumonia (PCP), which is common in infants with hyper IgM syndrome, is a serious illness. [9] PCP is one of the most frequent and severe opportunistic infections in people with weakened immune systems. ... "Hyper immunoglobulin M syndrome due to CD40 deficiency: clinical, molecular, and immunological features". ... "Update on the hyper immunoglobulin M syndromes" . British Journal of Haematology . 149 (2): 167–180. doi : 10.1111/j.1365-2141.2010.08077.x . ... Classification D ICD - 10 : D80.5 OMIM : 608106 External resources Orphanet : 101092 v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency
Description Hyper-IgM syndrome is a condition characterized by normal or increased serum IgM concentrations associated with low or absent serum IgG, IgA, and IgE concentrations, indicating a defect in the class-switch recombination (CSR) process (summary by Imai et al., 2003). ... Although the clinical manifestations were similar to those observed in HIGM2 (605258), which is caused by mutation in the gene encoding activation-induced cytidine deaminase (AICDA; 605257), these patients exhibited a slightly milder HIGM syndrome with residual IgG production. Imai et al. (2003) found that B-cell CSR was intrinsically impaired.
Primary immune deficiency disorder Hyper IgM syndrome type 5 Immunoglobulin M Symptoms Chronic diarrhea [1] Types Hyper-IgM syndrome type 1,2,3,4 and 5 [2] [3] [4] [5] [6] Diagnostic method MRI, Chest radiography and genetic testing [1] Treatment Allogeneic hematopoietic cell transplantation [7] The fifth type of hyper-IgM syndrome has been characterized in three patients from France and Japan. ... Contents 1 Hyper IgM syndromes 2 Signs and symptoms 3 Cause 4 Pathophysiology 5 Diagnosis 6 Treatment 7 References Hyper IgM syndromes [ edit ] Hyper IgM syndromes is a group of primary immune deficiency disorders characterized by defective CD40 signaling; via B cells affecting class switch recombination (CSR) and somatic hypermutation . ... As a consequence, people with HIGM have an increased susceptibility to infections. [9] [7] [10] Signs and symptoms [ edit ] Hyper IgM syndrome can have the following syndromes: [1] [11] Infection / Pneumocystis pneumonia (PCP), which is common in infants with hyper IgM syndrome, is a serious illness. [9] PCP is one of the most frequent and severe opportunistic infections in people with weakened immune systems. ... "Update on the hyper immunoglobulin M syndromes" . British Journal of Haematology . 149 (2): 167–180. doi : 10.1111/j.1365-2141.2010.08077.x . ... Classification D ICD - 10 : D80.5 OMIM : 608106 External resources Orphanet : 101092 v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency
A number sign (#) is used with this entry because autosomal recessive immunodeficiency with hyper-IgM type 5 (HIGM5) is caused by homozygous or compound heterozygous mutation in the gene encoding uracil-DNA glycosylase (UNG; 191525) on chromosome 12q23-q24.1. Description Hyper-IgM syndrome is a condition characterized by normal or increased serum IgM concentrations associated with low or absent serum IgG, IgA, and IgE concentrations, indicating a defect in the class-switch recombination (CSR) process.
Schimmelpenning syndrome Specialty Medical genetics Schimmelpenning syndrome is a neurocutaneous condition characterized by one or more sebaceous nevi, usually appearing on the face or scalp, [1] associated with anomalies of the central nervous system, ocular system, skeletal system, cardiovascular system and genitourinary system. [2] Synonyms include: " Linear nevus sebaceous syndrome (LNSS)", "Schimmelpenning-Feuerstein-Mims syndrome", "Feuerstein-Mims syndrome", "sebaceous nevus syndrome", "Solomon syndrome", and "Jadassohn's nevus phakomatosis". "Nevus" is sometimes spelled "naevus" and "sebaceous" may also be spelled "sebaceus". " Epidermal nevus syndrome " is sometimes used as a synonym, but more often as a broader term referring to Schimmelpenning syndrome in addition to nevus comedonicus syndrome , CHILD syndrome , Becker's nevus syndrome, and phakomatosis pigmentokeratotica . [3] The classic Schimmelpenning syndrome diagnosis comprises a triad of sebaceous nevi , seizures , and mental retardation . [2] The condition was first reported by Gustav Schimmelpenning in 1957 [4] and independently reported by Feuerstein and Mims in 1962. [5] Contents 1 Signs and symptoms 2 Genetic 3 Diagnosis 4 Management 5 Incidence 6 See also 7 References 8 External links Signs and symptoms [ edit ] Since the original identification of Schimmelpenning syndrome, the number of findings has expanded to the point that the syndrome is associated with a considerable constellation of abnormalities. [2] The abnormalities may occur in a variety of combinations, and need not include all three aspects of the classic triad of sebaceous nevus, seizures and mental retardation . ... It has been reported that up to 10% of individuals with epidermal nevi may develop additional syndrome symptoms, [3] but that number appears to be inconsistent with the rarity of the syndrome and may be overstated. [12] Prevalence is unknown, but Epidermal nevus syndrome is listed with the National Organization for Rare Disorders , which defines rare as affecting "fewer than 200,000 people in the United States." [13] See also [ edit ] Epidermis Epidermal nevus syndrome Skin lesion List of cutaneous conditions References [ edit ] ^ Menascu, Shay; Donner, Elizabeth J. (2008). "Linear nevus sebaceous syndrome: case reports and review of the literature". ... "Sebaceous lesions and their associated syndromes: Part I". J Am Acad Dermatol . 61 (4): 549–60. doi : 10.1016/j.jaad.2009.04.058 .
A rare nevus syndrome characterized by the association of an nevus sebaceous with a broad spectrum of abnormalities that affect many organ systems, most commonly the eye, skeletal and central nervous system. Epidemiology The incidence of epidermal nevus (EN) (isolated finding or as a syndrome) is estimated at 1-3/1,000 live births, with nevus sebaceous (NS) estimated at half of the total. Nevus sebaceous syndrome (NSS) is rare; the exact prevalence and incidence in the general population is unknown. ... Etiology NSS is a sporadic disease. EN syndromes are the result of post-zygotic mutations. ... Differential diagnosis Differential diagnosis includes cutaneous-skeletal hypophosphatemia syndrome, nevus comedonicus syndrome, Becker nevus syndrome, phakomatosis pigmentokeratotica, CHILD syndrome, the spectrum of PIK3CA-related overgrowth syndrome, SOLAMEN (segmental outgrowth-lipomatosis-arteriovenous malformation-epidermal nevus syndrome), and CLOVES syndrome.
Sandifer syndrome Other names Sandifer's syndrome Specialty Pediatrics Sandifer syndrome (or Sandifer's syndrome ) is an eponymous paediatric medical disorder, characterised by gastrointestinal symptoms and associated neurological features. [1] [2] [3] There is a significant correlation between the syndrome and gastro-oesophageal reflux disease (GORD); however, it is estimated to occur in less than 1% of children with reflux. [4] Contents 1 Symptoms and signs 2 Diagnosis 3 Treatment 4 Prognosis 5 History 6 See also 7 References 8 External links Symptoms and signs [ edit ] Onset is usually confined to infancy and early childhood, [2] with peak prevalence at 18–36 months. [4] In rare cases, particularly where the child is severely mentally impaired, onset may extend to adolescence. [4] The classical symptoms of the syndrome are spasmodic torticollis and dystonia . [3] [4] [5] Nodding and rotation of the head, neck extension, gurgling, writhing movements of the limbs, and severe hypotonia have also been noted. [3] Spasms may last for 1–3 minutes and may occur up to 10 times a day. ... Retrieved 11 December 2014 . ^ a b c d e "Orphanet: Sandifer syndrome" . Orpha.net . Retrieved 11 December 2014 . ^ a b c Theodoropoulos, D. ... F; Boyce, H W. (1999). "Sandifer's syndrome and gastro- oesophageal reflux disease" . ... PMID 17638161 . ^ Deskin, Ronald W. (1995). "Sandifer syndrome: A cause of torticollis in infancy". ... M.; Nalbantoglu, A (2013). "Sandifer's Syndrome: A Misdiagnosed and Mysterious Disorder" .
Sandifer syndrome is a paroxysmal dystonic movement disorder occurring in association with gastro-oesophageal reflux, and, in some cases, hiatal hernia. ... Although conflicting results have been obtained, some authors have suggested that the dystonic posture provides relief from abdominal pain. Diagnostic methods Sandifer syndrome is diagnosed on the basis of the association of gastro-oesophageal reflux with the characteristic movement disorder. ... Management and treatment Early diagnosis of the syndrome is essential, as effective treatment of the gastro-oesophageal reflux (by pharmacological therapy or surgical intervention) leads to resolution of the movement disorder.
Damiano Salpietro et al. (2004) reported a 10-year-old boy with what they considered to be Hallermann-Streiff syndrome (HSS; 234100). He was the second child of healthy, unrelated parents and showed short stature, a distinctive triangular face, a brachycephalic head with frontal bossing, sparse curly and thin scalp hair, hypoplastic eyebrows, and short, downslanted palpebral fissures with telecanthus and sparse eyelashes. ... Hennekam et al. (2010) stated that this patient 'clearly had oculo-dento-digital syndrome.' Molecular Genetics In 2 sisters with autosomal recessive ODDD, Richardson et al. (2006) identified homozygosity for a nonsense mutation in the GJA1 gene (121014.0016). In the patient described by Damiano Salpietro et al. (2004) as having Hallermann-Streiff syndrome but who was later determined to have ODDD (Hennekam et al., 2010), Pizzuti et al. (2004) identified a homozygous mutation in the GJA1 gene (121014.0017).
Cramer and Niederdellmann (1983) described 9 subjects from 3 families with cerebral gigantism syndrome (117550); 7 of the patients also had signs of the basal cell nevus syndrome. ... Goldstein et al. (1994) examined 11 African Americans from 2 families with Gorlin syndrome, which they abbreviated 'NBCC' for nevoid basal cell carcinoma syndrome. ... No patient had life-threatening sequelae of Gorlin-Goltz syndrome. The authors concluded that lifelong monitoring is essential for patient management in Gorlin-Goltz syndrome. ... Prenatal Diagnosis Bialer et al. (1994) made the prenatal diagnosis of Gorlin syndrome in a pregnancy sired by a man with Gorlin syndrome. ... History Although Gorlin has described many syndromes, several of which have been given his name, none is more intimately connected with his name than the basal cell nevus syndrome.
Oculodentodigital dysplasia is a condition that affects many parts of the body, including the eyes (oculo-), teeth (dento-), and fingers (digital). Symptoms of the condition include having small eyes, vision loss, missing teeth, frequent cavities, and bony growths in the fingers. The condition is caused by a mutation in GJA1 and is most typically inherited in an autosomal dominant manner. Oculodentodigital dysplasia can be diagnosed by a clinical examination and confirmed with genetic testing. Management is based on treating the specific symptoms that each affected person exhibits.
Oculodentodigital dysplasia is a condition that affects many parts of the body, particularly the eyes (oculo-), teeth (dento-), and fingers (digital). Common features in people with this condition are small eyes (microphthalmia) and other eye abnormalities that can lead to vision loss. Affected individuals also frequently have tooth abnormalities, such as small or missing teeth, weak enamel, multiple cavities, and early tooth loss. Other common features of this condition include a thin nose and webbing of the skin (syndactyly ) between the fourth and fifth fingers. Less common features of oculodentodigital dysplasia include sparse hair growth (hypotrichosis ), brittle nails, an unusual curvature of the fingers (camptodactyly ), syndactyly of the toes , small head size (microcephaly ), and an opening in the roof of the mouth (cleft palate ).
Genetic Heterogeneity of Oculodentodigital Syndrome An autosomal recessive form of ODDD (257850) is also caused by mutation in the GJA1 gene, but the majority of cases are autosomal dominant. ... Gillespie (1964) noted that similar features had been reported in 2 unrelated patients by Meyer-Schwickerath et al. (1957), who called the disorder oculodentodigital dysplasia and noted some phenotypic overlap with the Francois dyscephalic syndrome (Hallermann-Streiff syndrome; 234100). ... In 2 unpublished pedigrees, Renwick (1967) found that a constant and characteristic feature of the syndrome is the absence of the middle phalanx of those toes (second through fifth) that normally have 3 phalanges. ... Reisner et al. (1969) reported the syndrome in a mother and 3 of her 4 children. ... They reviewed other reports about neurologic defects in this syndrome and concluded that brain abnormalities may be a manifestation of the syndrome.
Mutations of the GJA1 gene have also been observed in syndactyly type 3 (see this term), suggesting that both syndromes are part of the same spectrum. ... Differential diagnosis Differential diagnosis includes a wide number of syndromes that present with skeletal, ocular, dental and neurological manifestations. ... Plastic or orthopedic surgery is indicated for severe limb malformations. Early recognition of the syndrome is of crucial importance in prevention and treatment of the wide variety of clinical manifestations.
Café au lait lesions with rough borders (“coast of Maine”) may be seen in McCune-Albright syndrome. [2] [3] In contrast, Café au lait lesions of neurofibromatosis have smooth borders (“coast of California"). [3] They are caused by a collection of pigment-producing melanocytes in the epidermis of the skin. [4] These spots are typically permanent and may grow or increase in number over time. [5] Café au lait spots are often harmless but may be associated with syndromes such as neurofibromatosis type 1 and McCune–Albright syndrome . [5] Contents 1 Cause 2 Diagnosis 3 Prognosis 4 Treatment 5 See also 6 References 7 External links Cause [ edit ] Neurofibromatosis type I café au lait spot Further information: List of conditions associated with café au lait macules Café au lait spots can arise from diverse and unrelated causes: [6] [7] Ataxia–telangiectasia Basal cell nevus syndrome Benign congenital skin lesion Bloom syndrome Chédiak–Higashi syndrome Congenital melanocytic naevus Fanconi anemia Gaucher disease Hunter syndrome Jaffe–Campanacci syndrome Legius syndrome Maffucci syndrome They can be caused by vitiligo in the rare McCune–Albright syndrome . [8] Multiple mucosal neuroma syndrome Having six or more café au lait spots greater than 5 mm in diameter before puberty, or greater than 15 mm in diameter after puberty, is a diagnostic feature of neurofibromatosis type I (NF-1), but other features are required to diagnose NF-1. [4] Familial multiple café au lait spots have been observed without an NF-1 diagnosis. [9] Noonan syndrome Silver–Russell syndrome Tuberous sclerosis Watson syndrome Wiskott–Aldrich syndrome Diagnosis [ edit ] Diagnosis is visual with measurement of spot size. ... However, they may be associated with syndromes such as Neurofibromatosis Type 1 and McCune-Albright syndrome . [5] The size and shape of the spots can vary in terms of description. ... "Café-au-lait spots caused by vitiligo in McCune-Albright syndrome" . J Bone Miner Res . 15 (12): 2521–2523. doi : 10.1359/jbmr.2000.15.12.2521 . ... Look up cafe au lait or macule in Wiktionary, the free dictionary. eMedicine v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation
It is the most common tumor of infancy . [8] PHACES Syndrome , a rare condition that often involves brain , heart , and arterial abnormalities, is generally accompanied by the presence of large facial hemangiomas. ... Often on the face, marks on the upper eyelid or forehead may be indicative of a condition called Sturge-Weber syndrome . Additionally, port-wine stains in these locations may be associated with glaucoma and seizures . [2] Treatment [ edit ] Most birthmarks are harmless and do not require treatment.
"Clinical and molecular cytogenetic characterisation of a newly recognised microdeletion syndrome involving 2p15-16.1" . J Med Genet . 44 (4): 269–76. doi : 10.1136/jmg.2006.045013 . ... "A newly recognised microdeletion syndrome of 2p15-16.1 manifesting moderate developmental delay, autistic behaviour, short stature, microcephaly, and dysmorphic features: a new patient with 3.2 Mb deletion" . ... "Further characterization of microdeletion syndrome involving 2p15-p16.1" . Am J Med Genet A . 152A (10): 2604–8. doi : 10.1002/ajmg.a.33612 . ... PMID 20799320 . ^ "2p15-16.1 microdeletion syndrome" . Wellcome Trust Sanger Institute . ... External links [ edit ] Classification D ICD - 10 : Q93.5 OMIM : 612513 MeSH : C567289 External resources Orphanet : 261349 DECIPHER database entry for 2p15-16.1 microdeletion syndrome Orphanet entry for 2p15-16.1 microdeletion syndrome Online Mendelian Inheritance in Man (OMIM): 612513 v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22 v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome (chr2:59.0-61.5 Mb; involving chromosome 2p16.1-p15). ... Chabchoub et al. (2008) reported a 16-year-old Belgian boy with 2p16.1-p15 deletion syndrome. He had mild mental retardation without autistic features. ... Liang et al. (2009) reported a 4.5-year-old Japanese girl with 2p16.1-p15 microdeletion syndrome. She showed intrauterine and postnatal growth retardation and severely delayed psychomotor development with an IQ of about 20. ... In an affected girl, Felix et al. (2010) was able to refine the critical region for the 2p16.1-p15 deletion syndrome to a 3.35-Mb region (chr2:59.13-62.48) that did not include the VRK2 gene. ... Molecular Genetics Funnell et al. (2015) concluded that increased HbF in patients with 2p16.1-p15 deletion syndrome is due to haploinsufficiency of the BCL11A gene.
2p15p16.1 microdeletion syndrome is a recently described syndrome characterized by developmental delay and facial dysmorphism. ... Microcephaly, short stature, genitourinary abnormalities and behavioral problems are common. Etiology This syndrome is caused by an interstitial deletion 2p15p16.1 (present in mosaic in one patient).
Shawl scrotum Specialty Urologist Shawl scrotum is a condition in which the scrotum surrounds the penis , resembling a ' shawl '. [1] It is a characteristic of some syndromes such as Aarskog-Scott syndrome (faciodigitogenital syndrome), Rubenstein-Taybi syndrome , craniofrontonasal dysplasia, Hunter Carpenter McDonald Syndrome , Naguib Syndrome, Saito Kuba Tsuruta Syndrome, Ieshima Koeda Inagaki syndrome, Cystic fibrosis Gastritis Megaloblastic Anemia, Willems de Vries syndrome , Schinzel syndrome and Seaver Cassidy syndrome . [ citation needed ] References [ edit ] ^ Reardon, William (2008).
Horner's syndrome Other names Bernard-Horner syndrome (BH), oculosympathetic palsy Left-sided Horner's syndrome Specialty Neurology Horner's syndrome , also known as oculosympathetic paresis, [1] is a combination of symptoms that arises when a group of nerves known as the sympathetic trunk is damaged. ... Diagnosis [ edit ] Left-sided Horner's syndrome in a cat as a result of trauma, demonstrating miosis in left pupil. ... However, in Horner's syndrome the lack of norepinephrine in the synaptic cleft causes mydriatic failure. ... In France and Italy , Claude Bernard is also eponymized with the condition (Claude Bernard–Horner syndrome, abbreviated CBH [10] ). In France, Francois Pourfour du Petit is also credited with describing this syndrome. ... "Neuroimaging strategies for three types of Horner syndrome with emphasis on anatomic location".
Overview Horner syndrome is a condition that affects the face and eye on one side of the body. It is caused by the disruption of a nerve pathway from the brain to the head and neck. Horner syndrome signs Decreased eye pupil size is a key sign of Horner syndrome. ... Horner syndrome is also known as Bernard-Horner syndrome or oculosympathetic palsy. Symptoms Horner syndrome usually affects only one side of the face. ... Tests to confirm Horner syndrome Your doctor may be able to diagnose Horner syndrome based on your history and an assessment of your symptoms.
Horner's syndrome is a rare condition characterized by miosis (constriction of the pupil), ptosis (drooping of the upper eyelid), and anhidrosis (absence of sweating of the face). ... The underlying causes of Horner's syndrome vary greatly and may include a tumor, stroke, injury, or underlying disease affecting the areas surrounding the sympathetic nerves. In rare cases, Horner's syndrome is congenital (present from birth) and may be associated with a lack of pigmentation of the iris (colored part of the eye). Treatment of Horner's syndrome depends on the underlying cause.
Some people are born with altered DNA that causes long QT syndrome (congenital long QT syndrome). ... Causes of congenital long QT syndrome More than a dozen genes have been linked to long QT syndrome so far. ... There are two forms of congenital long QT syndrome: Romano-Ward syndrome (autosomal dominant form). ... If a medication causes acquired long QT syndrome (LQTS), the condition may be called drug-induced long QT syndrome. ... It's important to understand that genetic tests for long QT syndrome can't detect all inherited cases of long QT syndrome.
In addition to the prolonged QT interval, associations include muscle weakness and facial dysmorphism in Andersen-Tawil syndrome (LQTS type 7); hand/foot, facial, and neurodevelopmental features in Timothy syndrome (LQTS type 8); and profound sensorineural hearing loss in Jervell and Lange-Nielson syndrome. ... Biallelic pathogenic variants in KCNQ1 are associated with Jervell and Lange-Nielsen syndrome. 8. The pathogenic variants in SCN5A that cause LQTS are gain-of-function variants (loss-of-function variants of SCN5A cause Brugada syndrome). ... LQTS type 8 is also referred to as Timothy syndrome. 7. Kimura et al [2012] 8. Biallelic pathogenic variants in KCNE1 are associated with Jervell and Lange-Nielsen syndrome. 9. ... LQTS type 7 is also referred to as Andersen-Tawil syndrome. 11. Shigemizu et al [2015] 12. ... Nomenclature The term "Romano-Ward syndrome" refers to forms of long QT syndrome with a purely cardiac phenotype, inherited in an autosomal dominant manner (LQTS types 1-3, type 5, type 6, and types 9-15).
Long QT syndrome ECG showing typical pattern of inherited Long QT syndrome (LQT1). ... Inherited [ edit ] Electrocardiograms from a single family showing unaffected family member (top), Romano Ward syndrome (middle) and Jervell and Lange-Nielsen syndrome (bottom). Inherited, or congenital long QT syndrome, is caused by genetic abnormalities. ... "Congenital long QT syndrome: considerations for primary care physicians". ... "How to perform and interpret provocative testing for the diagnosis of Brugada syndrome, long-QT syndrome, and catecholaminergic polymorphic ventricular tachycardia" .
A rare disorder characterized by periodic muscle paralysis, prolongation of the QT interval with a variety of ventricular arrhythmias (leading to predisposition to sudden cardiac death) and characteristic physical features: short stature, scoliosis, low-set ears, hypertelorism, broad nasal root, micrognathia, clinodactyly, brachydactyly and syndactyly. Etiology Mutations in KCNJ2 , which encodes the alpha subunit of the potassium channel Kir2.1, account for approximately 60% of cases. Genetic counseling AS is inherited as an autosomal dominant trait although sporadic cases have been reported. Penetrance is extremely variable. Management and treatment Treatment depends on the individual and their reaction to potassium. Patients with severe arrhythmias may require a pacemaker.
A number sign (#) is used with this entry because Andersen-Tawil syndrome is caused by heterozygous mutation in the KCNJ2 gene (600681) on chromosome 17q24. ... (This Andersen syndrome is not to be confused with Andersen disease, type IV glycogen storage disease (232500).) ... Tawil et al. (1994) showed that the Andersen syndrome is distinct from other forms of potassium-sensitive periodic paralysis by demonstrating lack of genetic linkage and concluded that it is probably distinct from the long QT syndrome (192500) on the same basis. ... Canun et al. (1999) suggested that recognition of the characteristic face in Andersen syndrome permits an early diagnosis and the detection of the severe systemic manifestations associated with the syndrome. ... Cleft palate was identified in 3 of 36 Andersen syndrome subjects (8%) and scoliosis in 4 of 36 (11%).
However, mild muscle weakness may eventually become permanent. In people with Andersen-Tawil syndrome, the most common changes affecting the heart are ventricular arrhythmia, which is a disruption in the rhythm of the heart's lower chambers (the ventricles), and long QT syndrome. Long QT syndrome is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. ... Physical abnormalities associated with Andersen-Tawil syndrome typically affect the face, other parts of the head, and the limbs. ... The signs and symptoms of Andersen-Tawil syndrome vary widely, and they can be different even among affected members of the same family. ... Researchers believe that Andersen-Tawil syndrome accounts for less than 10 percent of all cases of periodic paralysis.
Diagnosis Suggestive Findings Andersen-Tawil syndrome (ATS) should be suspected in individuals with either A or B: A. ... While the ECG may reveal a long QTc (LQT) interval, characteristic T-U patterns including enlarged U waves, a wide T-U junction, and prolonged terminal T-wave downslope distinguish ATS from other LQT syndromes [Zhang et al 2005, Haruna et al 2007]. ... Differential Diagnosis Andersen-Tawil syndrome (ATS) should be considered in any individual presenting with periodic paralysis and ventricular arrhythmias or prominent U wave or prolonged QTc. ... In one series a pathogenic variant in an inwardly rectifying potassium (Kir) channel (encoded by KCNJ18 ) was identified in approximately one third of affected individuals [Ryan et al 2010]. Long QT Syndromes See Long QT Syndrome, a review of similar phenotypes that are genetically diverse. ... Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with Andersen-Tawil syndrome (ATS), the evaluations summarized in Table 2 (if not performed as part of the evaluation that led to the diagnosis) are recommended: Table 2.
Andersen-Tawil syndrome is a type of long QT syndrome and is also considered a rare form of periodic paralysis . ... About 60% of cases of Andersen-Tawil syndrome are caused by mutations in the KCNJ2 gene.
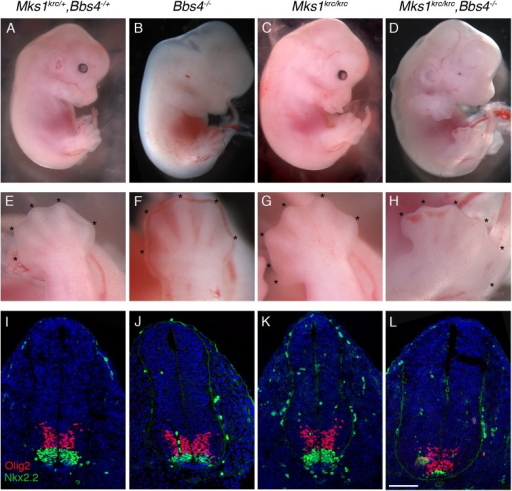
Prevention [ edit ] Prevention for Alström syndrome is considered to be harder compared to other diseases/syndromes because it is an inherited condition. ... Any person that has the syndrome have different set of disorders. ... Alström syndrome. GeneReviews. May 31, 2012; http://www.ncbi.nlm.nih.gov/books/NBK1267/ ^ "Alstrom Syndrome" . ... Retrieved 2015-12-06 . ^ a b c "Alström Syndrome — Symptoms, Diagnosis, Treatment of Alström Syndrome" . ... External links [ edit ] Classification D ICD - 10 : Q87.8 OMIM : 203800 606844 MeSH : D056769 DiseasesDB : 465 External resources MedlinePlus : 001665 GeneReviews : Alström Syndrome v t e Diseases of cilia Structural receptor: Polycystic kidney disease cargo: Asphyxiating thoracic dysplasia basal body : Bardet–Biedl syndrome mitotic spindle : Meckel syndrome centrosome : Joubert syndrome Signaling Nephronophthisis Other/ungrouped Alström syndrome Primary ciliary dyskinesia Senior–Løken syndrome Orofaciodigital syndrome 1 McKusick–Kaufman syndrome Autosomal recessive polycystic kidney See also: ciliary proteins
Charles et al. (1990) described Alstrom syndrome in the offspring of a couple related as first cousins once removed. ... This disorder is unusually frequent among French Acadians, both those living in Yarmouth County, Nova Scotia, and in Louisiana, where the syndrome may have been confused with Bardet-Biedl syndrome. ... The affected male became hypergonadal at a later age, consistent with late onset of components of the syndrome. Aynaci et al. (1995) described diabetes insipidus in association with Alstrom syndrome in a 16-year-old male patient. ... Awazu et al. (1997) found reports of 2 Japanese patients with Alstrom syndrome who had liver cirrhosis. Liver involvement in Alstrom syndrome appears to have been first described by Connolly et al. (1991). ... Lee et al. (2009) described an 18-month-old Taiwanese boy with Alstrom syndrome, in whom they identified a 19-bp deletion in exon 16 of the ALMS1 gene that had previously been found in another Taiwanese family with Alstrom syndrome (Marshall et al., 2007).
Alström syndrome is a rare condition that affects many body systems. ... The signs and symptoms of Alström syndrome vary in severity, and not all affected individuals have all of the characteristic features of the disorder. Frequency More than 900 people with Alström syndrome have been reported worldwide. Causes Mutations in the ALMS1 gene cause Alström syndrome. ... This protein is normally present at low levels in most tissues, so a loss of the protein's normal function may help explain why the signs and symptoms of Alström syndrome affect many parts of the body. Learn more about the gene associated with Alström syndrome ALMS1 Inheritance Pattern This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations.
Alström syndrome is a rare genetic disorder that affects many body systems. ... Over time, diabetes mellitus, liver problems, and slowly progressive kidney dysfunction which can lead to kidney failure may develop. Alström syndrome is caused by mutations in the ALMS1 gene. ... While there is no specific treatment for Alström syndrome, symptoms can be managed by a team of specialists with the goal of improving the quality of life and increasing the lifespan.
Genetic counseling. Alström syndrome is inherited in an autosomal recessive manner. ... The proportion of infants with Alström syndrome who develop infantile-onset cardiomyopathy is probably underestimated because some infants succumb to heart failure before the diagnosis of Alström syndrome is made. ... Of note, the proportion of those with Alström syndrome who develop infantile-onset cardiomyopathy may be underestimated because some infants may succumb before the diagnosis of Alström syndrome [Louw et al 2014]. ... Prevalence The prevalence of Alström syndrome is difficult to estimate; it is possible that individuals with attenuated forms of Alström syndrome may be underdiagnosed [Paisey et al 2011]. ... Disorders to Consider in the Differential Diagnosis of Alström Syndrome View in own window Disorder Gene(s) MOI Clinical Features Overlapping w/Alström syndrome Distinguishing from Alström syndrome (AS) Bardet-Biedl syndrome (BBS) >21 genes 1 AR 2 Rod-cone dystrophy Central obesity Hypogonadism Renal dysfunction Older mean age of onset of visual problems in BBS (8.5 yrs in BBS vs birth - 2 yrs in AS) Polydactyly is common in BBS (not described in AS).
A rare multisystemic disorder characterized by cone-rod dystrophy, hearing loss, obesity, insulin resistance and hyperinsulinemia, type 2 diabetes mellitus, dilated cardiomyopathy (DCM), and progressive hepatic and renal dysfunction. Epidemiology Alström syndrome (AS) has a suggested prevalence of 1/1000,000 in Europe and North America. ... Differential diagnosis Differential diagnoses include Bardet-Biedl syndrome, Biemond syndrome type 2, Wolfram syndrome, Cohen syndrome, familial isolated DCM and mitochondrial disorders.
Fetal trimethadione syndrome Other names German syndrome Condition is caused by Trimethadione (and paramethadione) Fetal trimethadione syndrome (also known as paramethadione syndrome , German syndrome , tridione syndrome , among others [1] ) is a set of birth defects caused by the administration of the anticonvulsants trimethadione (also known as Tridione) or paramethadione to epileptic mothers during pregnancy . [2] Fetal trimethadione syndrome is classified as a rare disease by the National Institute of Health 's Office of Rare Diseases, [3] meaning it affects less than 200,000 individuals in the United States . [4] The fetal loss rate while using trimethadione has been reported to be as high as 87%. [5] Contents 1 Presentation 2 Diagnosis 3 Treatment 4 References 5 External links Presentation [ edit ] Fetal trimethadione syndrome is characterized by the following major symptoms as a result of the teratogenic characteristics of trimethadione. [2] [6] Cranial and facial abnormalities which include; microcephaly , midfacial flattening, V-shaped eyebrows and a short nose Cardiovascular abnormalities Absent kidney and ureter Meningocele , a birth defect of the spine Omphalocele , a birth defect where portions of the abdominal contents project into the umbilical cord A delay in mental and physical development Listen to this article (1.2 megabytes) This audio file was created from a revision of this article dated 31 January 2007 ( 2007-01-31 ) , and does not reflect subsequent edits. ( Audio help · More spoken articles ) Diagnosis [ edit ] This section is empty. ... You can help by adding to it . ( July 2017 ) References [ edit ] ^ Additional names include trimethadione embryopathy and trimethadione syndrome . ^ a b Multiple Congenital Anomaly/Mental Retardation (MCA/MR) Syndromes - Retrieved January 2007 ^ Fetal trimethadione syndrome on the ORD website. Retrieved January 2007 ^ NIH's Office of Rare Diseases Archived 2009-02-06 at the Wayback Machine Retrieved January 2007 ^ Teratology and Drug Use During Pregnancy Retrieved January 2007 ^ The fetal trimethadione syndrome: report of an additional family and further delineation of this syndrome Retrieved January 2007 External links [ edit ] Classification D ICD - 10 : Q86.8 MeSH : C537798 External resources Orphanet : 1913
A drug-related embryofetopathy that can occur when an embryo/fetus is exposed to trimethadione and that is characterized by pre- and post-natal growth retardation, intellectual deficit, developmental and speech delay, craniofacial anomalies (with some similarities to those seen in fetal valproate syndrome), and less commonly, cleft palate, malformations of the heart, urogenital system and limbs.
Setleis syndrome Other names FFDD3 [1] Setleis syndrome is said to be inherited in an autosomal recessive manner. Specialty Dermatology Setleis syndrome is a very rare genetic condition characterized by facial skin abnormalities and double upper eyelashes and absent lower eyelashes. [2] It belongs to a group of diseases known as ectodermal dysplasias . Ectodermal dysplasias typically affect the hair , teeth, nails, and/or skin. Setleis syndrome is characterized by distinctive abnormalities of the facial area that may be apparent at birth ( congenital ). ... The differential diagnosis of Setleis syndrome includes X-linked focal dermal hypoplasia , or Goltz syndrome ; a syndrome of focal dermal hypoplasia, morning glory anomaly, and polymicrogyria; incontinentia pigmenti ; oculocerebrocutaneous syndrome ; Rothmund–Thomson syndrome ; and MLS (microphthalmia with linear skin defects) syndrome caused by deletions or point mutations in the HCCS gene. ... ISBN 978-1-4160-2999-1 . ^ "Setleis Syndrome" . External links [ edit ] Classification D ICD - 10 : Q82.8 OMIM : 227260 SNOMED CT : 403771007 External resources Orphanet : 1807 This dermatology article is a stub .
Clark et al. (1989) described 3 related children with Setleis syndrome who were not of Puerto Rican descent. ... Tsukahara et al. (1995) presented a follow-up on a Japanese patient with Setleis syndrome originally reported by Matsumoto et al. (1991). ... Inheritance Tukel et al. (2010) demonstrated that Setleis syndrome is an autosomal recessive disorder. ... Sequencing of TWIST2 in 5 additional unrelated probands with Setleis syndrome or focal facial dermal dysplasia did not reveal any disease-causing mutations, demonstrating genetic heterogeneity. In 2 Mexican-Nahua sibs with the characteristic features of Setleis syndrome, Cervantes-Barragan et al. (2011) identified homozygosity for a 1-bp deletion in the TWIST2 gene (607556.0003).
Focal facial dermal dysplasia type III (FFDD3) is a rare focal facial dermal dysplasia (FFDD; see this term), characterized primarily by congenital bitemporal scar-like depressions and a typical, but variable facial dysmorphism, which may include distichiasis (upper lids) or lacking eyelashes, slanted eyebrows and a flattened and/or bulbous nasal tip and other features such as a low frontal hairline, sparse hair, redundant skin, epicanthal folds, low-set dysplastic ears, blepharitis and conjunctivitis. Epidemiology FFDD3 is reported in over 20 patients from more than 15 families, but only 4 consanguineous families have had TWIST2 mutations. Clinical description FFDD3 is characterized by congenital bitemporal hypoplastic scar-like lesions resembling forceps marks with typical facial dysmorphic features. In addition, they may have periorbital puffiness (leonine facies), sparse lateral and upward lifting eyebrows, distichiasis (upper lashes), a lack of lower lashes and a prominent upper lip (with an inverted ''V'' contour). Nose abnormalities are very frequent and comprise a flattened and/or bulbous nasal tip with septum extended below the alae nasi.
This condition is also known as Brauer syndrome (hereditary symmetrical aplastic nevi of temples, bitemporal aplasia cutis congenita, bitemporal aplasia cutis congenita: OMIM 136500 ) and Setleis syndrome (facial ectodermal dysplasia: OMIM 227260 ). ... Other features that have been reported include dysplastic and low set ears, linear radiatory impressions on the forehead and congenital horizontal nystagmus . Those with the Setleis syndrome may be missing eyelashes on both the upper and lower lids or may have multiple rows of lashes on the upper lids but none on the lower lids. [ citation needed ] A possible association with intra abdominal cancer has been reported but to date this has not been confirmed in other studies. [1] Genetics [ edit ] Type II appears to be due to mutations in the transcription factor TWIST2 on chromosome 2 . [2] Type IV is due to mutations in the Cyp26c1 gene. [3] Pathology [ edit ] Under the temporal lesions the skeletal muscle is almost in direct continuity with the epidermis. Diagnosis [ edit ] Classification [ edit ] There are at least four types of FFDD: [4] Type I: autosomal dominant FFDD Type II: autosomal recessive FFDD Type III: FFDD with other facial features : Setleis syndrome [5] Type IV: facial lesions resembling aplasia cutis in a preauricular distribution along the line of fusion of the maxillary and mandibular prominences. ... You can help by adding to it . ( January 2017 ) History [ edit ] The syndrome was first described by Brauer in 1929 in a large five generation family (38 members). [6] The affected progenitor (Johann Jokeb Van Bargen) was a man who had migrated to Germany from Holland in the 16th century. ... "Nonsense mutations of the bHLH transcription factor TWIST2 found in Setleis Syndrome patients cause dysregulation of periostin" .
Clinical Features Ward and Moss (1994) suggested that the Setleis syndrome and type I focal facial dermal dysplasia (Brauer syndrome) 'are a single disorder.' ... Masuno et al. (1995) described a Japanese family in which a 9-month-old boy had typical Setleis syndrome; his father who had normal intelligence showed bitemporal focal dermal dysplasia but a normal face; and a paternal second cousin also had Setleis syndrome. McGaughran and Aftimos (2002) reported Setleis syndrome in 3 patients, a Caucasian boy and a father and son of Pacific Island descent. ... Father-to-son transmission of Setleis syndrome in the family reported by McGaughran and Aftimos (2002) was consistent with autosomal dominant inheritance. The transmission pattern of Brauer-Setleis syndrome in the families reported by Graul-Neumann et al. (2009) was consistent with autosomal dominant inheritance.
Focal facial dermal dysplasia type II (FFDD2) is a focal facial dermal dysplasia (FFDD; see this term), characterized by congenital bitemporal scar-like depressions and other facial and organ abnormalities. Epidemiology To date, FFDD2 has been reported in over 20 cases from 8 families. Clinical description FFDD2 is characterized by congenital bitemporal hypoplastic scar-like lesions resembling forceps marks with additional facial dysmorphic features. These frequently include low frontal hairline, sparse hair, periorbital puffiness, sparse lateral and upward lifting eyebrows, distichiasis (upper lashes), lack of lower lashes, flattened and/or bulbous nasal tip, and a prominent upper lip (with an inverted ''V'' contour). Occasionally epicanthal folds , linear grooves on forehead, skin dimples lateral to lips and redundant skin are reported.
Focal facial dermal dysplasia type I (FFDD1), also known as Brauer syndrome, is a focal facial dysplasia (FFDD; see this term) characterized by congenital bitemporal cutis aplasia.
Cervantes-Barragan et al. (2011) proposed a classification of FFDD in which there are 4 subtypes. FFDD1 (Brauer syndrome) is characterized by temporal skin depressions that resemble 'forceps marks.' ... Inheritance is autosomal dominant. FFDD3 (Setleis syndrome; 227260) is characterized by the same facial features as FFDD2, but the inheritance is autosomal recessive. ... Inheritance Father-to-son transmission was observed in the 3 large kindreds (German, English, Australian) with Brauer syndrome reported by Brauer (1929), Jensen (1971), and McGeoch and Reed (1971, 1973), indicating autosomal dominant inheritance. History Kowalski and Fenske (1992) proposed a classification in which Brauer syndrome was referred to as FFDD type I. They suggested that Setleis syndrome (focal facial dermal dysplasia with additional features) is a separate entity, which they classified as FFDD type III.