Full-field electroretinography (ERG) responses were undetectable to all stimulus conditions in all 4 sibs at ages ranging from 28 to 46 years, whereas an unaffected sib showed normal cone and rod responses at age 30 years.
"Ceruminous adenomas: a clinicopathologic study of 41 cases with a review of the literature". Am J Surg Pathol . 28 (3): 308–18. doi : 10.1097/00000478-200403000-00003 .
American Family Physician . 64 (3): 419–28. PMID 11515831 . Retrieved 2009-01-06 . ^ Cotran, Ramzi S.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005).
Such pathologies are arteriosclerosis or cerebral amyloid angiopathy. [2] Microinfarcts take longer to affect neuronal death progress, at up to 28 days, rather than hours. [3] Microinfarcts can range in size from 0.2 to 2.9 mm.
Clinical Features Chen et al. (2017) studied 2 sisters from a 4-generation Chinese family segregating autosomal dominant female-limited infertility. The proband was a 28-year-old woman who had normal ovarian reserves and regular menstrual cycles, with normal sex hormone levels and no abnormalities on infertility-related assessments.
Female infertility due to zona pellucida defect is a rare, genetic, female infertility disorder characterized by the presence of abnormal oocytes that lack a zona pellucida. Affected individuals are unable to conceive despite having normal menstrual cycles and sex hormone levels, as well as no obstructions in the fallopian tubes or defects of the uterus or adnexa.
A second major episode occurred during surgery performed when he was 28 years old. The third episode occurred during middle age when pulmonary bleeding led to hemoptysis.
P2Y12 defect is a rare hemorrhagic disorder characterized by mild to moderate bleeding diathesis with easy bruising, mucosal bleedings, and excessive post-operative hemorrhage due to defect of the platelet P2Y12 receptor resulting in selective impairment of platelet responses to adenosine diphosphate. Epidemiology To date, 14 patients have been described in the world literature. Clinical description P2Y12 defect is a congenital disorder that manifests by mildly to severely prolonged bleeding time, easy bruising, mucosal bleeding (epistaxis, gastric mucosa bleeding, gum bleeding, etc.), menorrhagia, and bleeding complications after trauma and minor or major surgery. Etiology P2Y12 defect is caused by mutations in the P2RY12 gene (3q24-q25) which result in the premature truncation of the P2Y12 receptor or in the synthesis of a dysfunctional P2Y12 receptor. ADP activates platelets through its interaction with two G protein-coupled receptors, P2Y1 and P2Y12.
Bleeding disorder due to P2RY12 defect affects the way the platelets function. Platelets are important for helping the blood to clot. Symptoms of a bleeding disorder due to P2RY12 defect include frequent nose bleeds, easy bruising, and excessive bleeding after surgery or an accident. These symptoms can vary from person to person. This condition is very rare and it's not clear how it changes over time. Bleeding disorder due to a P2RY12 defect occur due to a variant in the P2RY12 gene and is inherited in an autosomal recessive pattern. Diagnosis is based on the symptoms, clinical exam, and the results of specialized laboratory testing.
Molecular Genetics Sergouniotis et al. (2014) performed whole-exome sequencing in 28 patients with 'cone-first' retinal disease and clinical features that were atypical for ABCA4 (601691)-associated retinopathy (see CORD3, 604116).
INHERITANCE - Autosomal recessive HEAD & NECK Eyes - High myopia - Decreased visual acuity, rapidly progressive - Foveal hyperpigmentation - Foveal hyperfluorescence - Foveal atrophy (in some patients) - Defects in all axes of color vision - Central scotoma - Lack of photoreceptors in fovea by optical coherence tomography - Nondetectable photopic responses on electroretinography - Reduced scotopic responses on electroretinography MISCELLANEOUS - Onset of symptoms in the second decade of life MOLECULAR BASIS - Caused by mutation in the RAS-associated protein-28 gene (RAB28, 612994.0001 ) ▲ Close
Cone-rod dystrophy (CRD) is a group of inherited eye disorders that affect the light sensitive cells of the retina called the cones and rods . People with this condition experience vision loss over time as the cones and rods deteriorate. Initial signs and symptoms that usually occur in childhood may include decreased sharpness of vision (visual acuity) and abnormal sensitivity to light (photophobia). These signs are usually followed by blind spots in the central field of vision (scotomas), loss of color perception, and loss of peripheral vision. Most individuals with this condition are legally blind by mid adulthood.
For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy (CORD), see 120970. Clinical Features Khaliq et al. (2000) described a 2-generation, consanguineous Pakistani family with autosomal recessive cone-rod dystrophy. All affected individuals had night blindness, deterioration of central vision, photophobia, epiphora in bright light, and problems with color discrimination. Funduscopy revealed marked macular degeneration and attenuation of retinal vessels; mild pigmentary changes were present in the retinal periphery. Ismail et al. (2006) reexamined affected members of the Pakistani family with CORD8 previously described by Khaliq et al. (2000).
A number sign (#) is used with this entry because X-linked atrophic macular degeneration can be caused by mutation in the RPGR gene (312610). Clinical Features Ayyagari et al. (2002) described a family in which 10 males had primarily macular atrophy causing progressive loss of visual acuity with minimal peripheral visual impairment. One additional male showed extensive macular degeneration plus peripheral loss of retinal pigment epithelium and choriocapillaries. Full-field electroretinograms (ERGs) showed normal cone and rod responses in some affected males despite advanced macular degeneration. Mapping In a family with X-linked recessive atrophic macular degeneration, Ayyagari et al. (2002) mapped the disease locus to Xp21.1-p11.4, the region where the RPGR gene is situated.
For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy, see 120970. Clinical Features Kamenarova et al. (2013) studied a 3-generation Romani family segregating autosomal dominant cone-rod dystrophy with a slightly variable but early age of onset, at around 10 years of age. Affected individuals had gradual visual impairment and photophobia; ophthalmoscopy revealed typical signs of CORD including narrowing of retinal vessels, scattered bone-spicule pigmentation in the midperipheral retina, retinal pigment epithelium atrophy, and optic disc pallor. Electroretinography showed reduced photopic and scotopic responses, and visual field examination demonstrated central scotoma. Mapping In a 3-generation Romani family segregating autosomal dominant cone-rod dystrophy (adCORD), in which known loci associated with adCORD had been excluded, Kamenarova et al. (2013) performed a genomewide analysis using short tandem repeat markers and obtained a maximum lod score of 3.31 for a 6.7-Mb region on chromosome 10q26 between markers D10S1757 and D10S1782.
A number sign (#) is used with this entry because of evidence that cone-rod dystrophy-3 (CORD3) is caused by homozygous or compound heterozygous mutation in the ABCA4 (601691) on chromosome 1p22. For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy, see 120970. Clinical Features Klevering et al. (2002) analyzed phenotype information from the charts of 12 patients with autosomal recessive CORD caused by mutations in the ABCA4 gene and found that although the clinical presentation was heterogeneous, all patients experienced visual loss early in life, impaired color vision, and a central scotoma. Klevering et al. (2002) concluded that given the wide clinical spectrum of CORD-like phenotypes associated with ABCA4 mutations, detailed clinical subclassification is difficult and may not be very useful. Fishman et al. (2003) examined 30 patients with autosomal recessive CORD, 16 of whom harbored plausible disease-causing variations in the ABCA4 gene.
A number sign (#) is used with this entry because of evidence that cone-rod dystrophy-2 (CORD2) is caused by heterozygous mutation in the CRX gene (602225) on chromosome 19q13. Description Cone-rod dystrophy (CORD) characteristically leads to early impairment of vision. An initial loss of color vision and of visual acuity is followed by nyctalopia (night blindness) and loss of peripheral visual fields. In extreme cases, these progressive symptoms are accompanied by widespread, advancing retinal pigmentation and chorioretinal atrophy of the central and peripheral retina (Moore, 1992). In many families, perhaps a majority, central and peripheral chorioretinal atrophy is not found (Tzekov, 1998).
For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy (CORD), see 120970. Mapping Warburg et al. (1991) described a 20-year-old man with mental retardation and electrophysiologically demonstrated cone-rod dystrophy since childhood. He had hypogonadism and a central postsynaptic hearing impairment. Particularly noteworthy was the finding of deletion of the 18q21.1-qter segment. Three patients with more distal deletions on chromosome 18 did not present retinal dystrophies. This led Warburg et al. (1991) to suggest that a locus for cone-rod dystrophy may be located in the segment 18q21.1-q21.3.
A number sign (#) is used with this entry because of evidence that X-linked cone-rod dystrophy-3 (CORDX3) is caused by mutation in the CACNA1F gene (300110) on chromosome Xp11. Description Cone-rod dystrophy is a retinal disorder with predominantly cone involvement. Rod impairment may occur at the same time as the cone impairment or appear later. Patients with CORD usually have reduced visual acuity, photophobia, and color vision defects (summary by Huang et al., 2013). For a discussion of genetic heterogeneity of X-linked cone-rod dystrophy, see 304020.
A number sign (#) is used with this entry because of evidence that retinal dystrophy with early macular involvement (CORD21) is caused by homozygous or compound heterozygous mutation in the DRAM2 gene (613360) on chromosome 1p13. For a general phenotypic description and discussion of genetic heterogeneity of cone-rod dystrophy (CORD), see 120970. Clinical Features El-Asrag et al. (2015) studied a 5-generation consanguineous Pakistani family segregating autosomal recessive adult-onset retinal dystrophy with early macular involvement. Affected individuals described increasing difficulty with close visual tasks beginning in their thirties. There was progressive loss of visual acuity in all symptomatic individuals; light sensitivity and night blindness were inconsistent features of advanced disease.
A number sign (#) is used with this entry because of evidence that cone-rod dystrophy-9 (CORD9) is caused by homozygous or compound heterozygous mutation in the ADAM9 gene (602713) on chromosome 8p11. For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy, see 120970. Clinical Features Danciger et al. (2001) described a consanguineous Brazilian family segregating autosomal recessive CORD. Affected family members had childhood-onset visual acuity impairment, which progressed over decades to major loss of central and then peripheral visual function. In the 2 adult family members tested, visual acuity was 20/200 with a preserved midperipheral crescent on visual field testing.
A number sign (#) is used with this entry because of evidence that retinal cone dystrophy-4 (RCD4) is caused by homozygous mutation in the CACNA2D4 gene (608171) on chromosome 12p13. Clinical Features Wycisk et al. (2006) described 2 sibs with a recessive form of retinal cone dystrophy (RCD4). Symptoms were minimal except for slowly progressive reduction in visual acuity and moderate photophobia. Fundus examination showed nearly normal appearance in both. Color discrimination testing was consistent with defective color vision, and full-field electroretinography (ERG) showed moderately attenuated rod photoreceptor responses and markedly diminished cone responses. Although rod and cone ERGs suggested incomplete stationary night blindness, the authors noted that the disorder in these patients was progressive, not stationary, and concluded that their disease represented a mild cone dystrophy.
A number sign (#) is used with this entry because of evidence that cone-rod dystrophy-11 (CORD11) is caused by heterozygous mutation in the RAXL1 gene (RAX2, 610362) on chromosome 19p13. For a phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy, see 120970. Clinical Features Yang et al. (2015) studied a 4-generation family in which 6 members had retinal dystrophy. All affected individuals presented with declining visual acuity, although the age at onset of symptoms varied widely, with vision loss reported as early as age 15 years and as late as age 60. Examination of all 4 living patients revealed relative central scotoma on kinetic visual fields and obvious macular changes consistent with cone or cone-rod dystrophy, including small yellow macular deposits and/or macular pigment mottling, as well as waxy disc pallor and attenuated vasculature indicating diffuse dystrophy.
A number sign (#) is used with this entry because of evidence that cone-rod dystrophy-7 (CORD7) is caused by heterozygous mutation in the RIMS1 gene (606629) on chromosome 6q13. For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy, see 120970. Clinical Features Kelsell et al. (1998) described a 4-generation British family with autosomal dominant cone-rod dystrophy. Affected members first became aware of reduced color vision and visual acuity between the ages of 20 and 40 years. As the disorder progressed, they reported difficulty seeing in bright light.
A number sign (#) is used with this entry because X-linked cone-rod dystrophy-1 (CORDX1) and cone dystrophy-1 (COD1) are caused by mutation in an alternative terminal exon 15 (ORF15) of the RPGR gene (312610), which maps to chromosome Xp11. Description X-linked cone-rod dystrophy is a rare, progressive visual disorder primarily affecting cone photoreceptors (Demirci et al., 2002). Affected individuals, essentially all of whom are males, present with decreased visual acuity, myopia, photophobia, abnormal color vision, full peripheral visual fields, decreased photopic electroretinographic responses, and granularity of the macular retinal pigment epithelium. The degree of rod photoreceptor involvement is variable, with increasing degeneration. Although penetrance appears to be nearly 100%, there is variable expressivity with respect to age at onset, severity of symptoms, and findings (Hong et al., 1994).
Etiology Nonsyndromic CRDs are genetically heterogeneous (28 genes have been identified). The four most commonly mutated genes are ABCA4 (1p22.1) responsible for 30 to 60% of autosomal recessive CRDs, CRX (19q13.33) and GUCY2D (17p13.1) responsible for many reported cases of autosomal dominant CRDs, and RPGR (Xp11.4) responsible for X-linked CRDs.
A number sign (#) is used with this entry because of evidence that cone-rod dystrophy-10 (CORD10) can be caused by compound heterozygous mutation in the SEMA4A gene (607292) on chromosome 1q22. For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy, see 120970. Molecular Genetics Abid et al. (2006) screened 135 Pakistani patients with retinitis pigmentosa (RP35; 610282), 25 with cone-rod dystrophy, and 30 with congenital blindness for mutations in the SEMA4A gene. They identified compound heterozygosity for 2 substitutions (607292.0001-607292.0002) in 2 RP and 2 CORD patients. None of the mutations were found in 100 ethnically matched controls. INHERITANCE - Autosomal recessive HEAD & NECK Eyes - Loss of visual acuity, progressive - Loss of color vision, progressive - Night blindness - Loss of peripheral vision - Photophobia, severe - Epiphora, severe - Granular fundus - Macular degeneration - Retinal pigmentation, peripheral MISCELLANEOUS - Allelic with retinitis pigmentosa 35 ( 610282 ) MOLECULAR BASIS - Caused by mutation in the semaphorin 4A gene (SEMA4A, 607292.0001 ) ▲ Close
A number sign (#) is used with this entry because autosomal recessive cone-rod dystrophy-13 (CORD13) is caused by mutation in the RPGRIP1 gene (605446). For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy, see 120970. Clinical Features Hameed et al. (2003) reported 4 consanguineous Pakistani families with cone-rod dystrophy. Two of the families were large: 1 had 8 affected members over 2 generations, and the other had 8 affected members over 3 generations. The other 2 families had 2 affected sibs each. In all affected patients, there was deterioration in central vision and colorblindness from an early age, and rapid loss of vision between ages 14 and 16 years (visual acuity 1/60).
Police reportedly used a robot to examine it, [28] and a total of 300 rounds of ammunition were found inside. [29] Police officials warned of "an acute terror situation" and initially thought that there were up to three attackers, but later confirmed that there was only one gunman. [10] [30] The Munich U-Bahn , tram service, bus service, and services on the central portion of the S-Bahn in Munich were stopped. [31] [32] Munich main station was evacuated and all trains were cancelled in and out of Munich. ... Retrieved 23 July 2016 . ^ "Bayerische Polizei - Fortentwicklung der Ermittlungen zum Amoklauf in München" [Bavarian police - Evolution of the investigation into the massacre in Munich]. Polizei Bayern (in German). 28 July 2016 . Retrieved 1 August 2016 . ^ "Münchner Polizei bearbeitet rund 1750 Hinweise zu Amoklauf" [Munich police processed some 1,750 Notes on rampage]. Welt Online (in German). 28 July 2016 . Retrieved 1 August 2016 . ^ a b c d "David? ... Retrieved 30 August 2016 . ^ "Zentralrat Deutscher Sinti und Roma trauert um die Opfer des Münchner Anschlags – Drei Angehörige von Sinti und Roma unter den Opfern" [Central Council of German Sinti and Roma mourns the victims of the Munich attack - Three members of the Sinti and Roma among the victims]. Zentralrat (in German). 28 July 2016 . Retrieved 18 August 2016 . ^ "Lo sportivo e il piccolo eroe, chi erano le vittime di Monaco" [The sportsman and the little hero, who were the victims of Monaco]. ... The Economist . 27 July 2016 . Retrieved 28 July 2016 . ^ Chan, Rosalie (22 July 2016). "8 Dead in Munich Shooting" .
"The precursor of the H5N1 influenza virus that spread to humans in 1997 was first detected in Guangdong , China , in 1996, when it caused a moderate number of deaths in geese and attracted very little attention." [5] In 1997, in Hong Kong , 18 humans were infected and 6 died in the first known case of H5N1 infecting humans. [6] On December 28 to 29, 1997, 1.3 million chickens were killed by the government of Hong Kong. ... PMID 16494709 . ^ a b c WHO (October 28, 2005). "H5N1 avian influenza: timeline" (PDF) . ... February 27, 2008 . Retrieved February 28, 2008 . ^ "Avian influenza - situation in Egypt - update 5" . ... February 17, 2012. ^ "Cambodian girl, 10, dies from bird flu: WHO" . Google News Article . May 28, 2012. ^ "Speaking Notes – Deputy Chief Public Health Officer - H5N1 Technical Briefing" . ... Retrieved October 8, 2006 . ^ "German cat gets deadly bird flu" . BBC News. February 28, 2006 . Retrieved October 8, 2006 . ^ "Austrian Cats Test Positive for Bird Flu" .
The process may take several weeks. [3] Gently massaging the area around the blister can help loosen the worm. [9] This is nearly the same treatment that is noted in the famous ancient Egyptian medical text, the Ebers papyrus from c. 1550 BC. [10] Some people have said that extracting a Guinea worm feels as if the afflicted area is on fire. [26] [27] However, if the infection is identified before an ulcer forms, the worm can also be surgically removed by a trained doctor in a medical facility. [21] Although Guinea worm disease is usually not fatal, the wound where the worm emerges could develop a secondary bacterial infection such as tetanus , which may be life-threatening—a concern in endemic areas where there is typically limited or no access to health care. [28] Analgesics can be used to help reduce swelling and pain and antibiotic ointments can help prevent secondary infections at the wound site. [21] At least in the Northern region of Ghana, the Guinea worm team found that antibiotic ointment on the wound site caused the wound to heal too well and too quickly making it more difficult to extract the worm and more likely that pulling would break the worm. ... For many years the major focus was South Sudan (independent after 2011, formerly the southern region of Sudan ), which reported 76% of all cases in 2013. [31] In 2017 only Chad and Ethiopia had cases. [37] Date South Sudan Mali Ethiopia Chad Total 2011 1,028 [38] 12 [38] 8 [38] 10 [38] 1058 2012 521 [38] 7 [38] 4 [38] 10 [38] 542 2013 113 [38] 11 [38] 7 [38] 14 [38] 148 (including 3 exported to Sudan) 2014 70 [38] 40 [38] 3 [38] 13 [38] 126 2015 5 [38] 5 [38] 3 [38] 9 [38] 22 2016 6 [38] 0 [38] 3 [38] 16 [38] 25 2017 0 [34] 0 [34] 15 [34] 15 [34] 30 2018 10 [39] 0 [39] 0 [39] 17 [39] 28 (including one isolated case in Angola) 2019 4 [39] 0 [39] 0 [39] 47 [39] 53 (including one isolated case in Angola and Cameroon) Eradication program [ edit ] Main article: Eradication of dracunculiasis Logarithmic scale of reported human cases of guinea worm by year, 1989–2017 (2017 data is provisional). [40] Since humans are the principal host for Guinea worm, and there is no evidence that D. medinensis has ever been reintroduced to humans in any formerly endemic country as the result of non-human infections, the disease can be controlled by identifying all cases and modifying human behavior to prevent it from recurring. [9] [41] Over the years, the eradication program has faced several challenges: Inadequate security in some endemic countries Lack of political will from the leaders of some of the countries in which the disease is endemic The need for change in behaviour in the absence of a magic bullet treatment like a vaccine or medication Inadequate funding at certain times [11] The newly recognised transmission of guinea worm through non-human hosts (both domestic and wild animals) History [ edit ] Rod of Asclepius Dracunculiasis has been a recognized disease for thousands of years: Guinea worm has been found in calcified Egyptian mummies . [9] An Old Testament description of "fiery serpents" may have been referring to Guinea worm: "And the Lord sent fiery serpents among the people, and they bit the people; and much people of Israel died." ( Numbers 21:4–9). [10] In the 2nd century BC, the Greek writer Agatharchides described this affliction as being endemic amongst certain nomads in what is now Sudan and along the Red Sea. [42] [10] In the 18th century, Swedish naturalist Carl Linnaeus identified D. medinensis in merchants who traded along the Gulf of Guinea (West African Coast). ... "Why is dracunculiasis eradication taking so long?". Trends in Parasitology . 28 (6): 225–230. doi : 10.1016/j.pt.2012.03.003 . ... Retrieved 19 March 2014 . ^ "View Latest Worldwide Guinea Worm Case Totals" . www.cartercenter.org . Retrieved 28 February 2020 . ^ "Guinea Worm Cases Left in the World" . ... Archived from the original on 4 December 2014 . Retrieved 28 November 2014 . ^ Centers for Disease Control and, Prevention (25 October 2013).
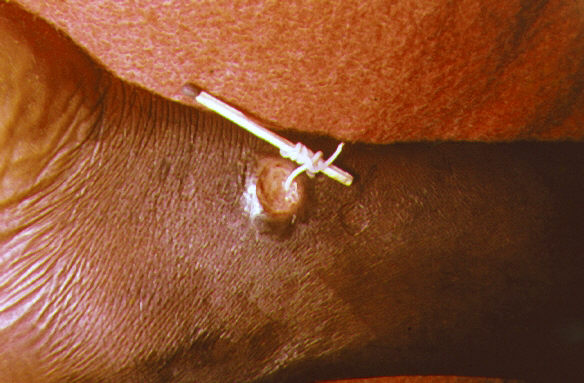
Dracunculiasis (Guinea worm disease) is a neglected tropical disease (NTD) characterized by a painful burning skin lesion from which the Dracunculus medinensis parasite emerges approximately 1 year after infection resulting from consumption of unsafe drinking water containing parasite-infected copepods ( Cyclops spp. , microcrustacea also called water fleas). Epidemiology In 2012, 542 cases were reported in 4 countries (Chad, Ethiopia, Mali, and South Sudan), a decrease of >99% since 1990. The global dracunculiasis program aims to eradicate the parasite from the last remaining endemic villages located in difficult to reach areas. Clinical description Clinical manifestations appear 10-14 months after infection and include constitutional symptoms (such as low-grade fever, itchy rash, nausea, vomiting, diarrhea, dizziness) followed by a localized swelling developing into a painful blister, most often on a lower limb. On contact with water, the adult female worm (70-100 cm) bursts through the blister, depositing her larvae in the water where they are consumed by copepods, starting the cycle anew.
Others Thyroid lymphoma Squamous cell thyroid carcinoma Sarcoma of thyroid Hürthle cell carcinoma The follicular and papillary types together can be classified as "differentiated thyroid cancer". [25] These types have a more favorable prognosis than the medullary and undifferentiated types. [26] Papillary microcarcinoma is a subset of papillary thyroid cancer defined as measuring less than or equal to 1 cm. [27] 43% of all thyroid cancers and 50% of new cases of papillary thyroid carcinoma are papillary microcarcinoma. [28] [29] Management strategies for incidental papillary microcarcinoma on ultrasound (and confirmed on FNAB) range from total thyroidectomy with radioactive iodine ablation to observation alone. ... "Thyroid and Parathyroid Cancers" Archived 28 February 2010 at the Wayback Machine in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management: A Multidisciplinary Approach Archived 4 October 2013 at the Wayback Machine . 11 ed. 2008. ^ Durante, Cosimo; Grani, Giorgio; Lamartina, Livia; Filetti, Sebastiano; Mandel, Susan J.; Cooper, David S. (6 March 2018). ... (Note:Book also states that the 14% 10-year survival for anaplastic thyroid cancer was overestimated) ^ Rounded up to nearest natural number from 96.7% as given by eMedicine Thyroid, Papillary Carcinoma Archived 28 July 2010 at the Wayback Machine Author: Luigi Santacroce. Coauthors: Silvia Gagliardi and Andrew Scott Kennedy. Updated: 28 September 2010 ^ By 100% minus cause-specific mortality of 17% at 5 yr, as given by Barbet J, Campion L, Kraeber-Bodéré F, Chatal JF (2005). ... Archived from the original on 17 October 2014 . Retrieved 28 October 2014 . ^ "Celebrities with Thyroid problems" .
Thyroid neoplasm Thyroid anatomy Specialty Oncology Thyroid neoplasm is a neoplasm or tumor of the thyroid . It can be a benign tumor such as thyroid adenoma , [1] or it can be a malignant neoplasm ( thyroid cancer ), such as papillary , follicular , medullary or anaplastic thyroid cancer . [2] Most patients are 25 to 65 years of age when first diagnosed; women are more affected than men. [2] [3] The estimated number of new cases of thyroid cancer in the United States in 2010 is 44,670 compared to only 1,690 deaths. [4] Of all thyroid nodules discovered, only about 5 percent are cancerous, and under 3 percent of those result in fatalities. Contents 1 Diagnosis 1.1 Classification 1.1.1 Benign neoplasms 1.1.2 Malignant neoplasms 2 Treatment 3 See also 4 References 5 External links Diagnosis [ edit ] Further information: Thyroid nodule The first step in diagnosing a thyroid neoplasm is a physical exam of the neck area. If any abnormalities exist, a doctor needs to be consulted. A family doctor may conduct blood tests, an ultrasound , and nuclear scan as steps to a diagnosis. The results from these tests are then read by an endocrinologist who will determine what problems the thyroid has.
HRT can be used with or without a progestogen to improve symptoms such as hot flashes, sweating, trouble sleeping, vaginal dryness and discomfort. [28] The FDA recommends HRT to be avoided in women with a history or risk of breast cancer, undiagnosed genital bleeding, untreated high blood pressure, unexplained blood clots, or liver disease. [28] HRT for the vasomotor symptoms of hypoestrogenism include different forms of estrogen, such as conjugated equine estrogens, 17β-estradiol, transdermal estradiol, ethinyl estradiol, and the estradiol ring. [28] In addition to HRT, there are common progestogens that are used to protect the inner layer of the uterus, the endometrium. These medications include medroxyprogesterone acetate , progesterone , norethisterone acetate , and drospirenone . [28] Non-pharmacological treatment of hot flashes includes using portable fans to lower the room temperature, wearing layered clothing, and avoiding tobacco, spicy food, alcohol and caffeine.
Supplementation with larger doses of amino acids, particularly leucine has been reported to counteract muscle loss with aging. [28] Exercise may work synergistically with amino acid supplementation. [18] β-hydroxy β-methylbutyrate (HMB) is a metabolite of leucine that acts as a signalling molecule to stimulate protein synthesis. [11] [18] It is reported to have multiple targets, including stimulating mTOR and decreasing proteasome expression. ... Biogerontology (Review). 9 (4): 213–28. doi : 10.1007/s10522-008-9131-0 . ... Journal of Cachexia, Sarcopenia and Muscle . 7 (1): 28–36. doi : 10.1002/jcsm.12048 . PMC 4799853 . ... PMID 26178029 . ^ a b c d Sakuma K, Yamaguchi A (28 May 2012). "Sarcopenia and age-related endocrine function" .
Lotions that help stop or prevent itching may be helpful. [26] [27] Direct sunlight makes the lesions resolve more quickly. [21] According to this principle, medical treatment with ultraviolet light has been used to hasten resolution, [28] though studies disagree whether it decreases itching [28] or not. [29] UV therapy is most beneficial in the first week of the eruption. [28] A 2007 meta-analysis concluded that there is insufficient evidence for the effectiveness of most treatments. [30] Oral erythromycin was found to be effective for treating the rash and relieving the itch based on one early trial; however, a later study could not confirm these results. [5] [30] [31] Prognosis [ edit ] In most patients, the condition lasts only a matter of weeks; in some cases it can last longer (up to six months). ... Annals of the Academy of Medicine, Singapore . 28 (6): 829–31. PMID 10672397 . ^ Trager, Jonathan D.
Overview Pityriasis rosea is a rash that often begins as an oval spot on the face, chest, abdomen or back. This is called a herald patch and may be up to 4 inches (10 centimeters) across. Then you may get smaller spots that sweep out from the middle of the body in a shape that looks like drooping pine-tree branches. The rash can be itchy. Pityriasis (pit-ih-RIE-uh-sis) rosea can happen at any age but is most common between the ages of 10 and 35. It tends to go away on its own within 10 weeks. Treatment may help relieve the symptoms.
Archived from the original on 13 May 2017 . Retrieved 28 May 2017 . ^ a b "Central Diabetes Insipidus" . ... Archived from the original on 21 February 2017 . Retrieved 28 May 2017 . ^ a b "Nephrogenic Diabetes Insipidus" . ... Archived from the original on 19 February 2017 . Retrieved 28 May 2017 . ^ a b Saborio P, Tipton GA, Chan JC (2000). ... Archived from the original on 2012-08-29 . Retrieved 2012-05-28 . ^ Perkins RM, Yuan CM, Welch PG (March 2006).
Overview Diabetes insipidus (die-uh-BEE-teze in-SIP-uh-dus) is an uncommon problem that causes the fluids in the body to become out of balance. That prompts the body to make large amounts of urine. It also causes a feeling of being very thirsty even after having something to drink. Diabetes insipidus also is called arginine vasopressin deficiency and arginine vasopressin resistance. While the terms "diabetes insipidus" and "diabetes mellitus" sound alike, the two conditions are not connected. Diabetes mellitus involves high blood sugar levels. It's a common condition, and it's often called simply diabetes.