-
Omenn Syndrome
Wikipedia
Omenn syndrome Omenn syndrome has an autosomal recessive pattern of inheritance. Specialty Hematology Omenn syndrome is an autosomal recessive severe combined immunodeficiency . [1] It is associated with hypomorphic missense mutations in immunologically relevant genes of T-cells (and B-cells ) such as recombination activating genes (RAG1 and RAG2), Interleukin-7 receptor-α (IL7Rα), DCLRE1C-Artemis , RMRP-CHH , DNA-Ligase IV , common gamma chain , WHN-FOXN1 , ZAP-70 and complete DiGeorge syndrome . ... Contents 1 Symptoms 2 Genetics 3 Diagnosis 4 Treatment 5 See also 6 References 7 External links Symptoms [ edit ] A 5-month-old female infant with Omenn syndrome; she has red, scaly skin all over her body. ... "The genetic and biochemical basis of Omenn syndrome". Immunol Rev . 178 : 64–74. doi : 10.1034/j.1600-065X.2000.17818.x . ... Department of Health and Human Services, National Institutes of Health, Genetic and Rare Diseases Information Center (last updated 2016). Omenn Syndrome. Retrieved from: https://rarediseases.info.nih.gov/diseases/8198/omenn-syndrome External links [ edit ] Classification D ICD - 10 : D81.2 ( ILDS D81.210) OMIM : 603554 DiseasesDB : 32676 External resources eMedicine : ped/1640 Orphanet : 39041 v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency

-
Leukocyte Adhesion Deficiency-1
Wikipedia
External links [ edit ] Classification D ICD - 10 : D84.8 OMIM : 116920 MeSH : C535887 DiseasesDB : 29751 External resources Orphanet : 99842 v t e Diseases of monocytes and granulocytes Monocytes and macrophages ↑ -cytosis : Monocytosis Histiocytosis Chronic granulomatous disease ↓ -penia : Monocytopenia Granulocytes ↑ -cytosis : granulocytosis Neutrophilia Eosinophilia / Hypereosinophilic syndrome Basophilia Bandemia ↓ -penia : Granulocytopenia/agranulocytosis ( Neutropenia / Severe congenital neutropenia / Cyclic neutropenia Eosinopenia Basopenia ) Disorder of phagocytosis Chemotaxis and degranulation Leukocyte adhesion deficiency LAD1 LAD2 Chédiak–Higashi syndrome Neutrophil-specific granule deficiency Respiratory burst Chronic granulomatous disease Neutrophil immunodeficiency syndrome Myeloperoxidase deficiency v t e Cell surface receptor deficiencies G protein-coupled receptor (including hormone ) Class A TSHR ( Congenital hypothyroidism 1 ) LHCGR ( Luteinizing hormone insensitivity , Leydig cell hypoplasia , Male-limited precocious puberty ) FSHR ( Follicle-stimulating hormone insensitivity , XX gonadal dysgenesis ) GnRHR ( Gonadotropin-releasing hormone insensitivity ) EDNRB ( ABCD syndrome , Waardenburg syndrome 4a , Hirschsprung's disease 2 ) AVPR2 ( Nephrogenic diabetes insipidus 1 ) PTGER2 ( Aspirin-induced asthma ) Class B PTH1R ( Jansen's metaphyseal chondrodysplasia ) Class C CASR ( Familial hypocalciuric hypercalcemia ) Class F FZD4 ( Familial exudative vitreoretinopathy 1 ) Enzyme-linked receptor (including growth factor ) RTK ROR2 ( Robinow syndrome ) FGFR1 ( Pfeiffer syndrome , KAL2 Kallmann syndrome ) FGFR2 ( Apert syndrome , Antley–Bixler syndrome , Pfeiffer syndrome , Crouzon syndrome , Jackson–Weiss syndrome ) FGFR3 ( Achondroplasia , Hypochondroplasia , Thanatophoric dysplasia , Muenke syndrome ) INSR ( Donohue syndrome Rabson–Mendenhall syndrome ) NTRK1 ( Congenital insensitivity to pain with anhidrosis ) KIT ( KIT Piebaldism , Gastrointestinal stromal tumor ) STPK AMHR2 ( Persistent Müllerian duct syndrome II ) TGF beta receptors : Endoglin / Alk-1 / SMAD4 ( Hereditary hemorrhagic telangiectasia ) TGFBR1 / TGFBR2 ( Loeys–Dietz syndrome ) GC GUCY2D ( Leber's congenital amaurosis 1 ) JAK-STAT Type I cytokine receptor : GH ( Laron syndrome ) CSF2RA ( Surfactant metabolism dysfunction 4 ) MPL ( Congenital amegakaryocytic thrombocytopenia ) TNF receptor TNFRSF1A ( TNF receptor associated periodic syndrome ) TNFRSF13B ( Selective immunoglobulin A deficiency 2 ) TNFRSF5 ( Hyper-IgM syndrome type 3 ) TNFRSF13C ( CVID4 ) TNFRSF13B ( CVID2 ) TNFRSF6 ( Autoimmune lymphoproliferative syndrome 1A ) Lipid receptor LRP : LRP2 ( Donnai–Barrow syndrome ) LRP4 ( Cenani–Lenz syndactylism ) LRP5 ( Worth syndrome , Familial exudative vitreoretinopathy 4 , Osteopetrosis 1 ) LDLR ( LDLR Familial hypercholesterolemia ) Other/ungrouped Immunoglobulin superfamily : AGM3, 6 Integrin : LAD1 Glanzmann's thrombasthenia Junctional epidermolysis bullosa with pyloric atresia EDAR ( EDAR hypohidrotic ectodermal dysplasia ) PTCH1 ( Nevoid basal-cell carcinoma syndrome ) BMPR1A ( BMPR1A juvenile polyposis syndrome ) IL2RG ( X-linked severe combined immunodeficiency ) See also cell surface receptorsITGB2, FERMT3, COL17A1, ITGAL, EGFR, LAD1, AKT1, MIR451A, MIR423, MIR326, PIK3CB, PIK3CA, PIK3CG, MIR214, UCN3, LINC00467, FOXP3, ITGAM, PIK3CD, AXL, HDAC1, CRNDE, SLC2A10, FEZF1-AS1, CKAP2L, KLRC4-KLRK1, RPLP0P2, CYTOR, SNHG12, HOXA10-AS, LINC-ROR, WNT5B, C5orf34, DANCR, TP73-AS1, MEG3, ANKRD40CL, TUG1, TERF2IP, GOLM1, ENKUR, SNHG5, MIR432, LINC01133, MIR650, CDKN2B-AS1, HOTAIR, MIR409, LINC01512, HOXA-AS3, MIR4270, LINC00673, RABGEF1, MIRLET7C, TTN-AS1, MIR29C, LINC00707, MIR21, MIR200B, MIR200A, MIR150, MIR144, CD274, KCNQ1OT1, IL37, HOXA10, PTEN, MAP2K1, MAPK1, NOTCH1, NM, KRAS, KDR, IL17A, CXCL8, IGHG3, HGF, ATP2C1, FGF2, EZH2, EPHB2, E2F3, DNMT1, DECR1, DDX3X, COL4A5, TNFRSF8, CASP1, RAC2, RAP1A, RET, SFRP1, SIRT1, WWC1, KLRK1, BIRC5, HCP5, DLC1, MTSS1, ATG12, SKAP2, HMGA2, XIST, VIM, VEGFA, VCAM1, TWIST1, TP53, THBS1, TEAD4, STK11, SP1, SOX5, MIR6839

-
Familial Exudative Vitreoretinopathy
Wikipedia
Familial exudative vitreoretinopathy Other names FEVR (initialism) , Criswick-Schepens syndrome Retina (located at top of diagram) Pronunciation (initialism) / ˈ f iː v ər / Specialty Ophthalmology Prevention treatment = laser surgery Familial exudative vitreoretinopathy ( FEVR , pronounced as fever ) is a genetic disorder affecting the growth and development of blood vessels in the retina of the eye. [1] This disease can lead to visual impairment and sometimes complete blindness in one or both eyes. ... External links [ edit ] Classification D ICD - 10 : H35.0 OMIM : 133780 MeSH : C580083 External resources Orphanet : 891 GeneReviews/NCBI/NIH/UW entry on Familial Exudative Vitreoretinopathy, Autosomal Dominant NCBI Genetic Testing Registry v t e Congenital malformations and deformations of eyes Adnexa Eyelid Ptosis Ectropion Entropion Distichia Blepharophimosis Ablepharon Marcus Gunn phenomenon Lacrimal apparatus Congenital lacrimal duct obstruction Globe Entire eye Anophthalmia ( Cystic eyeball , Cryptophthalmos ) Microphthalmia Lens Ectopia lentis Aphakia Iris Aniridia Anterior segment Axenfeld–Rieger syndrome Cornea Keratoglobus Megalocornea Other Buphthalmos Coloboma ( Coloboma of optic nerve ) Hydrophthalmos Norrie disease v t e Cell surface receptor deficiencies G protein-coupled receptor (including hormone ) Class A TSHR ( Congenital hypothyroidism 1 ) LHCGR ( Luteinizing hormone insensitivity , Leydig cell hypoplasia , Male-limited precocious puberty ) FSHR ( Follicle-stimulating hormone insensitivity , XX gonadal dysgenesis ) GnRHR ( Gonadotropin-releasing hormone insensitivity ) EDNRB ( ABCD syndrome , Waardenburg syndrome 4a , Hirschsprung's disease 2 ) AVPR2 ( Nephrogenic diabetes insipidus 1 ) PTGER2 ( Aspirin-induced asthma ) Class B PTH1R ( Jansen's metaphyseal chondrodysplasia ) Class C CASR ( Familial hypocalciuric hypercalcemia ) Class F FZD4 ( Familial exudative vitreoretinopathy 1 ) Enzyme-linked receptor (including growth factor ) RTK ROR2 ( Robinow syndrome ) FGFR1 ( Pfeiffer syndrome , KAL2 Kallmann syndrome ) FGFR2 ( Apert syndrome , Antley–Bixler syndrome , Pfeiffer syndrome , Crouzon syndrome , Jackson–Weiss syndrome ) FGFR3 ( Achondroplasia , Hypochondroplasia , Thanatophoric dysplasia , Muenke syndrome ) INSR ( Donohue syndrome Rabson–Mendenhall syndrome ) NTRK1 ( Congenital insensitivity to pain with anhidrosis ) KIT ( KIT Piebaldism , Gastrointestinal stromal tumor ) STPK AMHR2 ( Persistent Müllerian duct syndrome II ) TGF beta receptors : Endoglin / Alk-1 / SMAD4 ( Hereditary hemorrhagic telangiectasia ) TGFBR1 / TGFBR2 ( Loeys–Dietz syndrome ) GC GUCY2D ( Leber's congenital amaurosis 1 ) JAK-STAT Type I cytokine receptor : GH ( Laron syndrome ) CSF2RA ( Surfactant metabolism dysfunction 4 ) MPL ( Congenital amegakaryocytic thrombocytopenia ) TNF receptor TNFRSF1A ( TNF receptor associated periodic syndrome ) TNFRSF13B ( Selective immunoglobulin A deficiency 2 ) TNFRSF5 ( Hyper-IgM syndrome type 3 ) TNFRSF13C ( CVID4 ) TNFRSF13B ( CVID2 ) TNFRSF6 ( Autoimmune lymphoproliferative syndrome 1A ) Lipid receptor LRP : LRP2 ( Donnai–Barrow syndrome ) LRP4 ( Cenani–Lenz syndactylism ) LRP5 ( Worth syndrome , Familial exudative vitreoretinopathy 4 , Osteopetrosis 1 ) LDLR ( LDLR Familial hypercholesterolemia ) Other/ungrouped Immunoglobulin superfamily : AGM3, 6 Integrin : LAD1 Glanzmann's thrombasthenia Junctional epidermolysis bullosa with pyloric atresia EDAR ( EDAR hypohidrotic ectodermal dysplasia ) PTCH1 ( Nevoid basal-cell carcinoma syndrome ) BMPR1A ( BMPR1A juvenile polyposis syndrome ) IL2RG ( X-linked severe combined immunodeficiency ) See also cell surface receptors v t e Cell membrane protein disorders (other than Cell surface receptor , enzymes , and cytoskeleton ) Arrestin Oguchi disease 1 Myelin Pelizaeus–Merzbacher disease Dejerine–Sottas disease Charcot–Marie–Tooth disease 1B, 2J Pulmonary surfactant Surfactant metabolism dysfunction 1, 2 Cell adhesion molecule IgSF CAM : OFC7 Cadherin : DSG1 Striate palmoplantar keratoderma 1 DSG2 Arrhythmogenic right ventricular dysplasia 10 DSG4 LAH1 DSC2 Arrhythmogenic right ventricular dysplasia 11 Integrin : cell surface receptor deficiencies Tetraspanin TSPAN7 X-Linked mental retardation 58 TSPAN12 Familial exudative vitreoretinopathy 5 Other KIND1 Kindler syndrome HFE HFE hereditary haemochromatosis DYSF Distal muscular dystrophy Limb-girdle muscular dystrophy 2B See also other cell membrane proteinsLRP5, RCBTB1, ZNF408, CTNNB1, ABCA4, FZD4, TSPAN12, NDP, PRSS23, KIF11, FZD5, FZD1, MAOA, PLXNA2, JAG1, VEGFA, STK39, ILK, OPN1LW

-
Immunodeficiency–centromeric Instability–facial Anomalies Syndrome
Wikipedia
Find sources: "Immunodeficiency–centromeric instability–facial anomalies syndrome" – news · newspapers · books · scholar · JSTOR ( August 2008 ) ( Learn how and when to remove this template message ) ICF syndrome Other names Immunodeficiency-centromeric instability-facial anomalies syndrome ICF syndrome (or I mmunodeficiency, C entromere instability and F acial anomalies syndrome ) [1] is a very rare autosomal recessive [2] immune disorder . ... "ICF syndrome (immunodeficiency, centromeric instability and facial anomalies): investigation of heterochromatin abnormalities and review of clinical outcome". ... "DNMT3B mutations and DNA methylation defect define two types of ICF syndrome". Human Mutation . 25 (1): 56–63. doi : 10.1002/humu.20113 . ... "Hematopoietic Stem Cell Transplantation Corrects the Immunologic Abnormalities Associated with Immunodeficiency Centromeric Instability Facial Dysmorphism Syndrome". Pediatrics . 120 (5): e1341–e1344. doi : 10.1542/peds.2007-0640 . PMID 17908720 . ^ https://www.ptglab.com/news/blog/icf-syndrome-a-gene-silencing-chromatin-disorder/ IFC Syndrome: A gene silencing chromatin disorder External links [ edit ] Classification D ICD - 10 : D84.8 OMIM : 242860 MeSH : C537362 DiseasesDB : 32366 External resources Orphanet : 2268 Orphanet Journal of Rare Diseases link to ICF syndrome [1] v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency This immunology article is a stub .
-
Klippel-Feil Syndrome
Medlineplus
Less than half of all individuals with Klippel-Feil syndrome have all three classic features of this condition. ... People with Klippel-Feil syndrome may have a wide variety of other features in addition to their spine abnormalities. ... Rarely, structural brain abnormalities or a type of birth defect that occurs during the development of the brain and spinal cord (neural tube defect) can occur in people with Klippel-Feil syndrome. In some cases, Klippel-Feil syndrome occurs as a feature of another disorder or syndrome, such as Wildervanck syndrome or hemifacial microsomia. In these instances, affected individuals have the signs and symptoms of both Klippel-Feil syndrome and the additional disorder. Frequency Klippel-Feil syndrome is estimated to occur in 1 in 40,000 to 42,000 newborns worldwide. ... As a feature of another disorder, Klippel-Feil syndrome is inherited in whatever pattern the other disorder follows.
-
Lennox-Gastaut Syndrome
Medlineplus
Lennox-Gastaut syndrome is a severe condition characterized by recurrent seizures (epilepsy) that begin early in life. ... Other types of seizures have been reported less frequently in people with Lennox-Gastaut syndrome. Seizures associated with Lennox-Gastaut syndrome often do not respond well to therapy with anti-epileptic medications. ... Most children with Lennox-Gastaut syndrome have intellectual disability or learning problems even before seizures begin. ... For unknown reasons, it appears to be more common in males than in females. Causes Lennox-Gastaut syndrome can have many different causes. ... Many people with Lennox-Gastaut syndrome have a history of epilepsy beginning in infancy (infantile spasms) or a related condition called West syndrome before developing the features of Lennox-Gastaut syndrome.
-
Naxos Syndrome
Wikipedia
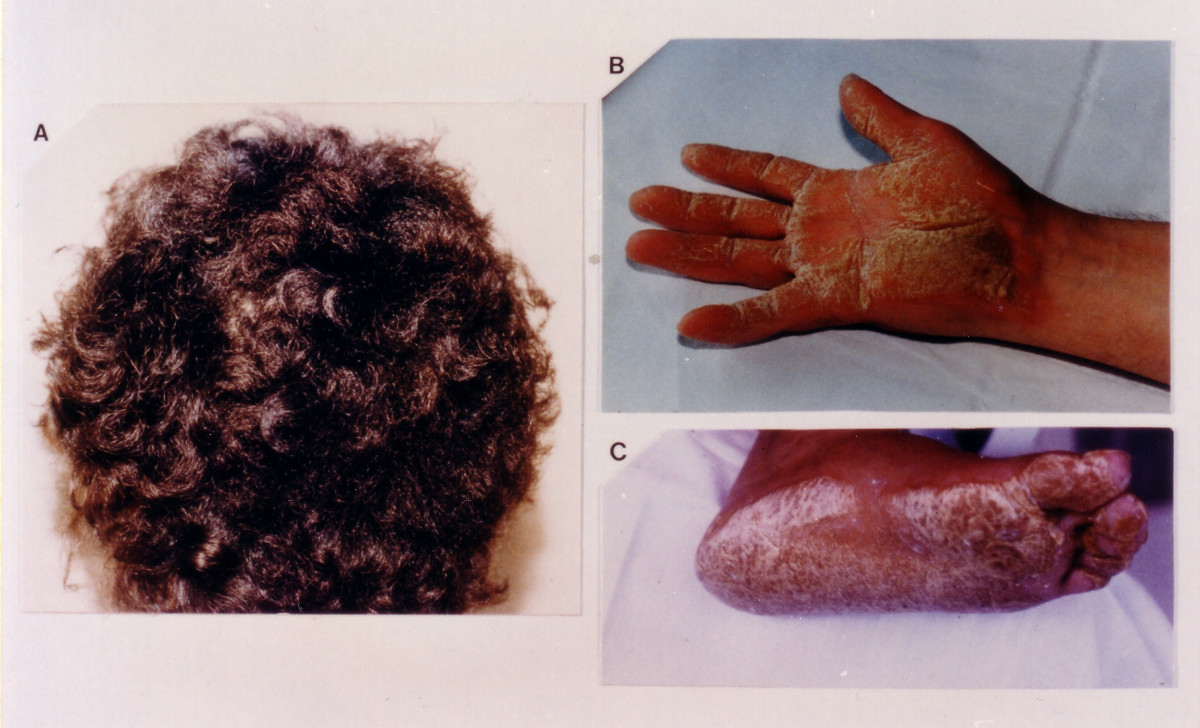
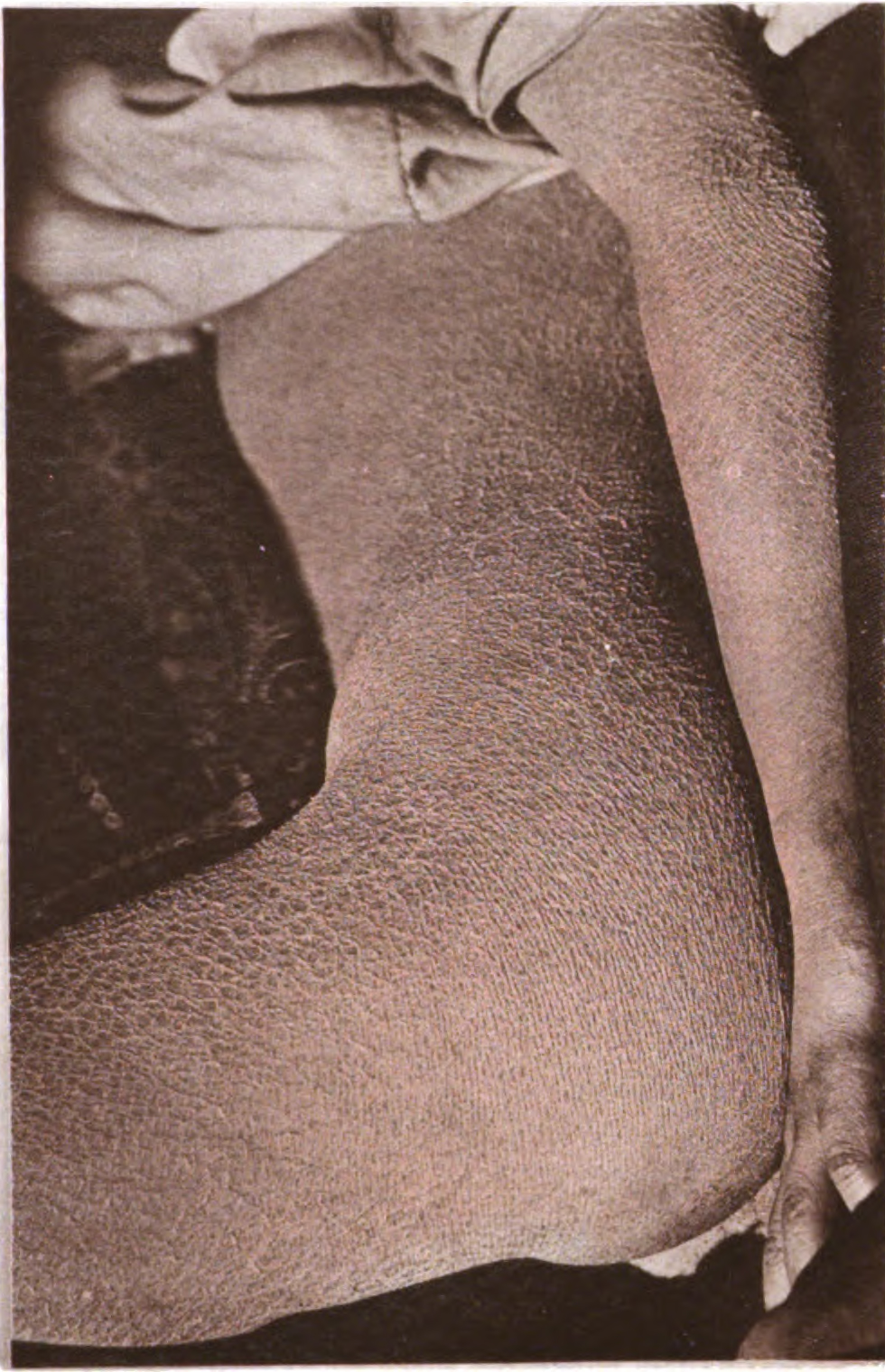
Naxos disease (also known as "Diffuse non-epidermolytic palmoplantar keratoderma with woolly hair and cardiomyopathy," [1] "Diffuse palmoplantar keratoderma with woolly hair and arrhythmogenic right ventricular cardiomyopathy firstly described in Naxos island by Dr Nikos Protonotarios," [1] and "Naxos disease" [1] ) is a cutaneous condition characterized by a palmoplantar keratoderma . [1] The prevalence of the syndrome is up to 1 in every 1000 people in the Greek islands. [2] It has been associated with mutations in the genes encoding the proteins desmoplakin , plakoglobin , desmocollin-2 , and SRC-interacting protein (SIP) . [3] [4] A variation of Naxos syndrome is known as Carvajal syndrome. [2] See also [ edit ] Olmsted syndrome List of cutaneous conditions List of conditions caused by problems with junctional proteins References [ edit ] ^ a b c d Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... "Naxos disease: Cardiocutaneous syndrome due to cell adhesion defect" . ... External links [ edit ] Classification D OMIM : 601214 MeSH : C538346 v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins This dermatology article is a stub .
-
Kaufman Oculocerebrofacial Syndrome
Wikipedia
Kaufman oculocerebrofacial syndrome Other names Blepharophimosis-ptosis-intellectual disability syndrome Kaufman oculocerebrofacial syndrome has an autosomal recessive pattern of inheritance . ... Additionally to ascertain if the individual has the condition: [3] [4] Growth assessment Thyroid function evaluation Kidney ultrasound Echocardiogram Differential diagnosis [ edit ] Kaufman oculocerebrofacial syndrome differential diagnosis consists of: [3] Ohdo syndrome Smith–Lemli–Opitz syndrome Maat–Kievit–Brunner syndrome Chromosome 3pter-p25 deletion syndrome Management [ edit ] Treatment for this condition entails surveillance of growth and contractures . ... Retrieved 2017-07-29 . ^ a b "OMIM Entry - # 244450 - KAUFMAN OCULOCEREBROFACIAL SYNDROME; KOS" . omim.org . Retrieved 21 October 2017 . ^ a b c d e f Basel-Vanagaite, Lina; Borck, Guntram (1993). ... PMID 27763745 . update 2016 ^ a b "Kaufman Oculocerebrofacial Syndrome, Sequencing UBE3B Gene - Tests - GTR - NCBI" . www.ncbi.nlm.nih.gov . Retrieved 21 October 2017 . ^ Kaufman R, Rimoin D, Prensky A, Sly W (1971). "An oculocerebrofacial syndrome". Birth Defects Orig Artic Ser . 7 (1): 135–138.
-
Young–simpson Syndrome
Wikipedia
(May 1999). "Young-Simpson syndrome: further delineation of a distinct syndrome with congenital hypothyroidism, congenital heart defects, facial dysmorphism, and mental retardation". ... "Young-Simpson syndrome comprising transient hypothyroidism, normal growth, macular degeneration and torticolis". ... "A Japanese boy with Young-Simpson syndrome". Acta Paediatr Jpn . 39 (4): 472–4. doi : 10.1111/j.1442-200x.1997.tb03621.x . ... PMID 10955481 . ^ OHDO SYNDROME: Contact a Family - for families with disabled children: information on rare syndromes and disorders ^ a b Bonthron DT, Barlow KM, Burt AM, Barr DG (March 1993). "Parental consanguinity in the blepharophimosis, heart defect, hypothyroidism, mental retardation syndrome (Young-Simpson syndrome)" . J Med Genet . 30 (3): 255–6. doi : 10.1136/jmg.30.3.255 .

-
Edwards Syndrome
Wikipedia
Archived from the original on 2016-10-02. ^ "Edwards syndrome (John Hilton Edwards)" . WhoNamedIt.com. ... PMID 17206726 . ^ Imataka, George; Yamanouchi, Hideo; Arisaka, Osamu (2007). "Dandy–Walker syndrome and chromosomal abnormalities". ... "The Most Common Comorbidities in Dandy-Walker Syndrome Patients: A Systematic Review of Case Reports" (PDF) . ... Retrieved 15 January 2021 . ^ "Prevalence and Incidence of Edwards Syndrome" . Diseases Center-Edwards Syndrome . ... Retrieved 2008-02-17 . mean maternal age for this disorder is 32½ External links [ edit ] Classification D ICD - 10 : Q91.0 - Q91.3 ICD - 9-CM : 758.2 MeSH : D000073842 DiseasesDB : 13378 External resources MedlinePlus : 001661 eMedicine : ped/652 Patient UK : Edwards syndrome Orphanet : 3380 Edwards syndrome at Curlie Perinatal Hospice Care - Preparing for birth and death" Humpath #5389 v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22 Authority control GND : 1046751034 NDL : 01161959AFP, PAPPA, BCL2, IGH, BCL6, MALT1, ADAM12, AFA, TTC3, PGF, SERPINB5, RPL17, TERC, TP53, VAPA, ZFY, PAEP, CKAP2, LGALS13, CHD7, CNDP2, RETN, CGB5, CGB8, SERPINB2, MYC, NOTCH1, DCC, ANPEP, BIRC3, APOA1, APOE, CD38, CDC25C, CGA, CGB3, FPR1, NEUROD2, FUT1, GH1, GHR, HTC2, IGHG3, KCNMA1, CD200, MTHFR, APCDD1

-
Ichthyosis
Wikipedia
Ichthyosis vulgaris accounts for more than 95% of cases. [4] Contents 1 Types 1.1 Non-Syndromic Ichthyosis 1.2 Syndromic Ichthyosis 1.3 Non-genetic ichthyosis 2 Diagnosis 3 Treatments 4 Other animals 5 See also 6 References 7 External links Types [ edit ] Many types of ichthyoses exist, and an exact diagnosis may be difficult. Types of ichthyoses are classified by their appearance, if they are syndromic or not, and by mode of inheritance. [5] For example, non-syndromic ichthyoses that are inherited recessively come under the umbrella term autosomal recessive congenital ichthyosis (ARCI). ... Of note, X-linked ichthyosis is associated with Kallmann syndrome (close to the KAL1 gene ). The most common or well-known types are: [5] Non-Syndromic Ichthyosis [ edit ] Name OMIM Mode Of Inheritance Gene(s) Ichthyosis Vulgaris 146700 Autosomal semi-dominant FLG X-linked recessive ichthyosis 308100 X-linked recessive STS Harlequin ichthyosis 242500 Autosomal recessive ABCA12 Congenital ichthyosiform erythoderma 242100 Autosomal recessive TGMI1 , NIPAL4 , ALOX12B , ALOXE3 , ABCA12 , CYP4F22 , NIPAL4, LIPN, CERS3, PNPLA1, ST14, CASP14 Lamellar ichthyosis 242300 Autosomal recessive TGMI1 , NIPAL4 , ALOX12B , ALOXE3 , ABCA12 , CYP4F22 , NIPAL4, LIPN, CERS3, PNPLA1, ST14, CASP14 Self improving congenital ichthyosis 242300 Autosomal recessive TGM1 , ALOX12B , ALOXE3 Bathing suit ichthyosis 242300 Autosomal recessive TGMI1 Epidermolytic ichthyosis 113800 Autosomal dominant KRT1 , KRT10 Superficial epidermolytic ichthyosis 146800 Autosomal dominant KRT2 Annular epidermolytic ichthyosis 607602 Autosomal dominant KRT1 , KRT10 Ichthyosis Curth-Macklin 146590 Autosomal dominant KRT1 Autosomal recessive epidermolytic ichthyosis 113800 Autosomal recessive KRT10 Congenital reticular ichthyosiform erythroderma 609165 Autosomal dominant KRT1 , KRT10 Epidermolytic nevi 113800 Postzygotic mosaicism KRT1 , KRT10 Loricrin keratoderma 604117 Autosomal dominant LOR Erythrokeratodermia variabilis 133200 Autosomal dominant GJB3 , GJB4 Peeling skin disease 270300 Autosomal recessive CDSN Keratosis linearis with ichthyosis congenita and sclerosing keratoderma 601952 Autosomal recessive POMP Syndromic Ichthyosis [ edit ] Name OMIM Mode Of Inheritance Gene(s) X-linked recessive ichthyosis syndromic forms 308700 300500 300533 X-linked recessive STS Ichthyosis follicularis with alopecia and photophobia syndrome 308205 X-linked recessive MBTPS2 Conradi-Hunermann-Happle syndrome 302960 X-linked dominant EBP Netherton syndrome 256500 Autosomal recessive SPINK5 Ichthyosis-hypotrichosis syndrome 610765 Autosomal recessive ST14 Trichothiodystrophy 601675 Autosomal recessive ERCC2 , ERCC3 , GTF2H5 Trichothiodystrophy (non-congenital forms) 275550 211390 601675 Autosomal recessive C7Orf11 , TTDN1 Sjögren-Larsson syndrome 270200 Autosomal recessive ALDH3A2 Refsum's disease 266500 Autosomal recessive PHYH , PEX7 Mental retardation, enteropathy, deafness, neuropathy, ichthyosis, keratoderma syndrome 609528 Autosomal recessive SNAP29 Arthrogryposis, renal dysfunction, cholestasis syndrome 208085 Autosomal recessive VPS33B Keratitis-ichthosis-deafness syndrome 602450 148210 Autosomal dominant GJB2 Neutral lipid storage disease with ichthyosis 275630 Autosomal recessive ABHD5 Ichthyosis prematurity syndrome 608649 Autosomal recessive SLC27A4 Non-genetic ichthyosis [ edit ] Ichthyosis acquisita Diagnosis [ edit ] A physician often can diagnose ichthyosis by looking at the skin. ... One of which is limb reduction defect known as CHILD syndrome ; a rare inborn error of metabolism of cholesterol biosynthesis that is usually restricted to one side of the body. ... DermAtlas 1896838546 Ichthyosis Overview - US National Institute of Arthritis and Musculoskeletal and Skin Diseases v t e Diseases of the skin and appendages by morphology Growths Epidermal Wart Callus Seborrheic keratosis Acrochordon Molluscum contagiosum Actinic keratosis Squamous-cell carcinoma Basal-cell carcinoma Merkel-cell carcinoma Nevus sebaceous Trichoepithelioma Pigmented Freckles Lentigo Melasma Nevus Melanoma Dermal and subcutaneous Epidermal inclusion cyst Hemangioma Dermatofibroma (benign fibrous histiocytoma) Keloid Lipoma Neurofibroma Xanthoma Kaposi's sarcoma Infantile digital fibromatosis Granular cell tumor Leiomyoma Lymphangioma circumscriptum Myxoid cyst Rashes With epidermal involvement Eczematous Contact dermatitis Atopic dermatitis Seborrheic dermatitis Stasis dermatitis Lichen simplex chronicus Darier's disease Glucagonoma syndrome Langerhans cell histiocytosis Lichen sclerosus Pemphigus foliaceus Wiskott–Aldrich syndrome Zinc deficiency Scaling Psoriasis Tinea ( Corporis Cruris Pedis Manuum Faciei ) Pityriasis rosea Secondary syphilis Mycosis fungoides Systemic lupus erythematosus Pityriasis rubra pilaris Parapsoriasis Ichthyosis Blistering Herpes simplex Herpes zoster Varicella Bullous impetigo Acute contact dermatitis Pemphigus vulgaris Bullous pemphigoid Dermatitis herpetiformis Porphyria cutanea tarda Epidermolysis bullosa simplex Papular Scabies Insect bite reactions Lichen planus Miliaria Keratosis pilaris Lichen spinulosus Transient acantholytic dermatosis Lichen nitidus Pityriasis lichenoides et varioliformis acuta Pustular Acne vulgaris Acne rosacea Folliculitis Impetigo Candidiasis Gonococcemia Dermatophyte Coccidioidomycosis Subcorneal pustular dermatosis Hypopigmented Tinea versicolor Vitiligo Pityriasis alba Postinflammatory hyperpigmentation Tuberous sclerosis Idiopathic guttate hypomelanosis Leprosy Hypopigmented mycosis fungoides Without epidermal involvement Red Blanchable Erythema Generalized Drug eruptions Viral exanthems Toxic erythema Systemic lupus erythematosus Localized Cellulitis Abscess Boil Erythema nodosum Carcinoid syndrome Fixed drug eruption Specialized Urticaria Erythema ( Multiforme Migrans Gyratum repens Annulare centrifugum Ab igne ) Nonblanchable Purpura Macular Thrombocytopenic purpura Actinic/solar purpura Papular Disseminated intravascular coagulation Vasculitis Indurated Scleroderma / morphea Granuloma annulare Lichen sclerosis et atrophicus Necrobiosis lipoidica Miscellaneous disorders Ulcers Hair Telogen effluvium Androgenic alopecia Alopecia areata Systemic lupus erythematosus Tinea capitis Loose anagen syndrome Lichen planopilaris Folliculitis decalvans Acne keloidalis nuchae Nail Onychomycosis Psoriasis Paronychia Ingrown nail Mucous membrane Aphthous stomatitis Oral candidiasis Lichen planus Leukoplakia Pemphigus vulgaris Mucous membrane pemphigoid Cicatricial pemphigoid Herpesvirus Coxsackievirus Syphilis Systemic histoplasmosis Squamous-cell carcinoma v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation BirthmarkPNPLA1, GJB2, SGPL1, SUPV3L1, MDM2, STS, FLG, TGM1, ABCA12, ST14, ALDH3A2, ALOX12B, VPS33B, SNAP29, ALOXE3, SLC27A4, ELOVL4, NIPAL4, CYP4F22, TGM5, CERS3, EBP, SDR9C7, AP1S1, MPDU1, ANOS1, KRT14, MBTPS2, CLDN1, KRT16, PEX11B, MAP2K1, FGF17, NEK9, HESX1, NLRP3, MAP2K2, PEX3, SRP54, PEX19, PEX2, RNF113A, PEX5, MSMO1, TARS1, TACR3, SOX10, SULT2B1, STIM1, PEX10, SEMA3A, HS6ST1, SRD5A3, PROK2, CARD14, SLURP1, TMEM43, WDR11, PEX26, CHD7, SLC29A3, IL17RD, EFL1, POMP, SBDS, ABHD5, SPRY4, PEX16, PSAT1, PHGDH, NSMF, FLRT3, SYNE1, SYNE2, DOLK, KISS1R, GJB6, PHYH, GINS1, ORAI1, PIGL, PROKR2, VIPAS39, PEX14, EMD, CARMIL2, PEX13, IL2RB, TMPRSS6, HRAS, CCDC141, GTF2E2, GBA, SUMF1, FHL1, FGFR1, FGF8, ERCC3, ERCC2, DUSP6, KRT1, FLG-AS1, DCC, CSTA, FEZF1, COL7A1, COL4A5, CHKB, BRAF, BLM, GTF2H5, ARSL, LIPN, SNHG31, AHSG, KRAS, ITGB6, KRT2, ND5, KRT6A, TRNH, TRNL1, TRNQ, TRNS1, TRNS2, TRNW, MPLKIP, NRAS, DNAJC21, PEX1, PEX6, PEX7, NOD2, PEX12, ND6, TRNF, ND4, LMNA, KRT6B, KRT17, COX1, ND1, COX2, COX3, PNPLA2, SPINK5, IL17A, KIF22, IL23A, AQP6, LBR, KRT10, FITM2, ASPRV1, IL17F, ELOVL3, DMKN, SLC27A1, ABCA3, SULT2A1, ERVK-6, FMR1, JUN, IFNG, HSPB2, HSPB1, HLA-DOA, GATA3, DSP, NLGN4X, DMD, CST6, CLDN7, BGN, ARSA, AMY2B, MX1, SLC26A4, PLIN1, AP1B1, TNF, TXNRD1, PUDP, HSD17B6, DGAT1, HSPB3, LPIN2, PNPLA6, IL36B, IL22, GAL, VCX3A, IL36G, ERVK-32
-
Epidermolysis Bullosa Simplex
Wikipedia
Associated with a recurrent mutation in KRT14. [3] : 557 [4] [5] 12q13 ( KRT5 ) 131960 Epidermolysis bullosa simplex, autosomal recessive 17q12-q21 ( KRT14 ) 601001 Generalized epidermolysis bullosa simplex Also known as "Koebner variant of generalized epidermolysis bullosa simplex", presents at birth to early infancy with a predilection for the hands, feet, and extremities, and palmar-plantar hyperkeratosis and erosions may be present. [1] : 598 [3] : 556 17q12-q21 ( KRT5 ), 12q13 ( KRT14 ) 131900 Localized epidermolysis bullosa simplex Also known as "Weber–Cockayne syndrome", [5] : 460 and "Weber–Cockayne variant of generalized epidermolysis bullosa simplex", is characterized by onset in childhood or later in life, and is the most common variant of epidermolysis bullosa simplex. [1] : 598 [3] : 557 17q12-q21 ( KRT5 ), 17q11-qter, 12q13 ( KRT14 ) 131800 Epidermolysis bullosa herpetiformis Also known as "Dowling-Meara epidermolysis bullosa simplex", presents at birth with a generalized distribution, often with oral mucosa involvement and variable lesions in infancy. [1] : 598 [3] : 557 17q12-q21 ( KRT5 ), 12q13 ( KRT14 ) 131760 Epidermolysis bullosa simplex with muscular dystrophy A rare clinical entity, and is the only epidermolytic epidermolysis bullosa described that is not caused by a keratin mutation, presenting as a generalized intraepidermal blistering similar to the Koebner variant of generalized epidermolysis bullosa simplex , but also associated with adult onset muscular dystrophy. [1] : 598 [3] : 557 [5] 8q24 ( PLEC1 ) 226670 Epidermolysis bullosa simplex with pyloric atresia 8q24 ( PLEC1 ) 612138 Epidermolysis bullosa simplex of Ogna Has onset in infancy, presenting with seasonal blistering on acral areas during summer months. [1] : 598 [3] : 557 [5] 8q24 ( PLEC1 ) 131950 Management [ edit ] No cure for EB Treat symptoms Protect skin, stop blister formation, promote healing Prevent complications Necessary treatment: use oral and topical steroid for healing and prevent complication Maintain cool environment, avoid overheating and decreases friction See also [ edit ] Epidermolysis bullosa List of cutaneous conditions caused by mutations in keratins References [ edit ] ^ a b c d e f Freedberg, et al. (2003). ... GeneReviews/NCBI/UW/NIH entry on Epidermolysis Bullosa Simplex External links [ edit ] Classification D ICD - 10 : Q81.0 ICD - 9-CM : 757.39 OMIM : 131900 131760 131800 131960 MeSH : D016110 DiseasesDB : 4334 External resources eMedicine : derm/124 Orphanet : 304 v t e Diseases of the skin and appendages by morphology Growths Epidermal Wart Callus Seborrheic keratosis Acrochordon Molluscum contagiosum Actinic keratosis Squamous-cell carcinoma Basal-cell carcinoma Merkel-cell carcinoma Nevus sebaceous Trichoepithelioma Pigmented Freckles Lentigo Melasma Nevus Melanoma Dermal and subcutaneous Epidermal inclusion cyst Hemangioma Dermatofibroma (benign fibrous histiocytoma) Keloid Lipoma Neurofibroma Xanthoma Kaposi's sarcoma Infantile digital fibromatosis Granular cell tumor Leiomyoma Lymphangioma circumscriptum Myxoid cyst Rashes With epidermal involvement Eczematous Contact dermatitis Atopic dermatitis Seborrheic dermatitis Stasis dermatitis Lichen simplex chronicus Darier's disease Glucagonoma syndrome Langerhans cell histiocytosis Lichen sclerosus Pemphigus foliaceus Wiskott–Aldrich syndrome Zinc deficiency Scaling Psoriasis Tinea ( Corporis Cruris Pedis Manuum Faciei ) Pityriasis rosea Secondary syphilis Mycosis fungoides Systemic lupus erythematosus Pityriasis rubra pilaris Parapsoriasis Ichthyosis Blistering Herpes simplex Herpes zoster Varicella Bullous impetigo Acute contact dermatitis Pemphigus vulgaris Bullous pemphigoid Dermatitis herpetiformis Porphyria cutanea tarda Epidermolysis bullosa simplex Papular Scabies Insect bite reactions Lichen planus Miliaria Keratosis pilaris Lichen spinulosus Transient acantholytic dermatosis Lichen nitidus Pityriasis lichenoides et varioliformis acuta Pustular Acne vulgaris Acne rosacea Folliculitis Impetigo Candidiasis Gonococcemia Dermatophyte Coccidioidomycosis Subcorneal pustular dermatosis Hypopigmented Tinea versicolor Vitiligo Pityriasis alba Postinflammatory hyperpigmentation Tuberous sclerosis Idiopathic guttate hypomelanosis Leprosy Hypopigmented mycosis fungoides Without epidermal involvement Red Blanchable Erythema Generalized Drug eruptions Viral exanthems Toxic erythema Systemic lupus erythematosus Localized Cellulitis Abscess Boil Erythema nodosum Carcinoid syndrome Fixed drug eruption Specialized Urticaria Erythema ( Multiforme Migrans Gyratum repens Annulare centrifugum Ab igne ) Nonblanchable Purpura Macular Thrombocytopenic purpura Actinic/solar purpura Papular Disseminated intravascular coagulation Vasculitis Indurated Scleroderma / morphea Granuloma annulare Lichen sclerosis et atrophicus Necrobiosis lipoidica Miscellaneous disorders Ulcers Hair Telogen effluvium Androgenic alopecia Alopecia areata Systemic lupus erythematosus Tinea capitis Loose anagen syndrome Lichen planopilaris Folliculitis decalvans Acne keloidalis nuchae Nail Onychomycosis Psoriasis Paronychia Ingrown nail Mucous membrane Aphthous stomatitis Oral candidiasis Lichen planus Leukoplakia Pemphigus vulgaris Mucous membrane pemphigoid Cicatricial pemphigoid Herpesvirus Coxsackievirus Syphilis Systemic histoplasmosis Squamous-cell carcinoma v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteinsKRT5, KRT14, EXPH5, DST, PLEC, ITGB4, NAT9, COL17A1, KLHL24, KRT1, VIM, TNF, MMP9, PLOD3, MAPK8, TGM5, CPSF4, ACTB, KRT10, KRT17, KRT9, IVL, CXCL8, IL6, IL2, HSP90AA1, GFAP, FN1, DES, BHLHE23

-
Dilated Cardiomyopathy With Ataxia Syndrome
Medlineplus
A small percentage of people with DCMA syndrome have no heart problems at all. By age 2, children with DCMA syndrome have problems with coordination and balance (ataxia). These movement problems can result in delay of motor skills such as standing and walking, but most older children with DCMA syndrome can walk without support. In addition to heart problems and movement difficulties, most individuals with DCMA syndrome grow slowly before and after birth, which leads to short stature. ... DCMA syndrome is associated with increased levels of a substance called 3-methylglutaconic acid in the urine. ... People with DCMA syndrome also have high urine levels of another acid called 3-methylglutaric acid.
-
Peho Syndrome
Wikipedia
PEHO syndrome Other names Progressive encephalopathy with edema, hypsarrhythmia and optic atrophy Usual onset Infancy, Neonatal Diagnostic method Mainly clinical, MRI Differential diagnosis Aicardi syndrome, mevalonic aciduria, CDG syndromes, autosomal recessive cerebellar hypoplasia, Joubert syndrome, olivo-pontine cerebellar atrophies Treatment Supportive care Prognosis Very poor; most die before age 15 PEHO syndrome (Progressive encephalopathy with Edema, Hypsarrhythmia and Optic atrophy) is an autosomal recessive and dominate, progressive neurodegenerative disorder that starts in the first few weeks or months of life. ... Other features include arrest of global developmental delay , severe intellectual deficit , encephalopathy , tapered fingers, and facial dysmorphism. [1] There is no specific treatment for PEHO syndrome; only the symptoms associated with the syndrome can be managed. ... "PEHO syndrome" . ^ Vanhatalo S, Somer M, Barth PG (April 2002). ... (September 2003). "PEHO and PEHO-like syndromes: report of five Australian cases". ... "The phenotypic and molecular spectrum of PEHO syndrome and PEHO-like disorders" . Brain . 140 (8): e49. doi : 10.1093/brain/awx155 .
-
Sjögren Syndrome
Medlineplus
Sjögren syndrome is a disorder whose main features are dry eyes and a dry mouth. ... In all, about half of all individuals with Sjögren syndrome also have another autoimmune disorder. Frequency Sjögren syndrome is a relatively common disorder; it occurs in 0.1 to 4 percent of the population. ... Causes Sjögren syndrome is thought to result from a combination of genetic and environmental factors; however, no associations between specific genetic changes and the development of Sjögren syndrome have been confirmed. ... Relatives of people with Sjögren syndrome are at an increased risk of developing autoimmune diseases, although they are not necessarily more likely to develop Sjögren syndrome in particular.STAT4, TNFAIP3, TNIP1, GTF2I, IL12A, FCGR2A, NCF1, PTTG1, ITSN2, PHIP, TNF, PRDM1, IRAK1BP1, DGKQ, TNFSF13B, THBS1, IL2, IL2RA, IL4, TGFB1, NFKBIA, CCR7, CYP19A1, AIRE, ID3, ST14, FAS, TRAF3IP2, MAP3K14, HMOX1, E2F1, RORC, IRF5, HLA-DPB1, RELN, BLK, IL10, IL12A-AS1, KLRG1, COL11A2P1, TRIM21, HLA-DRA, CALR, ATN1, RO60, SSB, IL17A, IFNG, TNPO3, SHISA9, IL6, AQP5, IL1B, CXCL13, CTSS, BPIFA2, HLA-DRB1, CXCL10, LILRA3, MIR146A, RBM45, CA6, MUC1, HMGB1, BCL2, EGF, IL18, IL23A, CXCL12, CXCL8, IFNA1, HT, MMP9, IL21, IL22, REG1A, MYDGF, MUC5AC, ISG20, IL13, ICAM1, IFNB1, IFNA13, OAS1, HLA-A, RTEL1, CRISP3, TXLNA, VCAM1, LINC02605, TRAF6, TRBV20OR9-2, CNTN2, TNFSF13, CXCR5, BMP6, SPTAN1, CASP3, ADIPOQ, CCL21, ACR, CHRM3, IL7, NLRP3, IGHV3-52, NGF, PRL, MYD88, STXBP3, SYT1, ADAM17, TAP2, CCR9, XBP1, PSPN, TP53, MALT1, PUF60, TXN, BTG3, VEGFA, VIP, IL17D, STAT1, TLR7, LTA, RAB4A, PSPH, ITPR3, ITPR1, MBL2, ITGB2, PSMB9, PTEN, ITGAL, ITGAE, MSMB, MDK, TREX1, RIDA, CCL18, CXCL9, CXCL11, TRIM38, FOXP3, PAX6, RBBP4, IL27, IL17F, GZMA, GSTT1, GSTM1, LOC102723407, CD40, AQP1, GSN, CXCL1, HLA-DQA1, AQP4, AREG, ASCL2, CXCR3, IL33, C3, MTOR, HLA-B, EGFR, LOC102724971, CTLA4, IL5, AGER, IL1A, ALB, IL23R, ICA1, IGH, HRES1, CDKN2A, MUC19, MIR200B, ABCA7, RAB3D, CLOCK, APOBEC3B, MIR31, IKBKE, MIR223, LINC01139, MFN2, NR1I3, MIR181A2, CHST3, MIR130A, AKT3, EBI3, PRAMEF13, PRG4, NCR3, MIR155, CDK2AP2, TRIM22, ATG5, UGT2B28, ABCG2, MIR34C, H3P8, XDH, LAPTM5, CXCR4, H3P13, TAM, SEMA7A, PERCC1, USO1, TP63, IFNG-AS1, VAMP8, MIR4484, MIR4695, TNFSF14, OCLN, IL18R1, MIR1207, MIR1248, NRP1, PSS, SEMA3A, MIR551B, MIR483, MIR146B, MIR448, MIR377, MSC, MIAT, MUC5B, SLC26A9, NOD1, NAPB, TRBV5-6, TRBV6-2, PDLIM3, CHIA, AICDA, EDA2R, ACAD8, NUDT10, HEYL, GNL3, MYCBP, ATRNL1, ROBO3, DDX58, LAMTOR2, PYCARD, TBK1, CD274, RETN, ATF7IP, VAC14, RNPC3, TRAT1, LEF1, IL17RB, NUDT15, NAT10, TRIM68, SF3B6, RNF125, TET2, GORASP1, PADI4, IL25, TPPP, IFNLR1, NXF1, NRSN1, TMEM132D, TNFRSF13C, DNAJC10, GRAP, DBA2, CCL27, SUB1, MUC16, TMED2, CKAP4, DSTN, RPP14, SEC14L2, WDHD1, CHP1, NT5C1A, HMCN1, NR1I4, FAM167A, ATF6, MAP1LC3B, CUL9, ASRGL1, KCNH3, ARHGAP45, DHX40, WNK1, XBP1P1, ABCA1, TRPV1, VIPR2, GOLGA4, GOLGA1, FUCA2, FUCA1, FPR2, FMR1, FLNB, F9, ESR1, ERV3-1, EPHB2, ELN, EDNRA, EDA, DNASE1, DLAT, CYP1A1, CX3CR1, CTSD, GOT2, GPT, GRN, CCN1, TNFRSF9, IL12RB1, CXCR2, CXCR1, IL7R, IL6R, IL1RN, RBPJ, IFI27, GRIN2A, IFI16, HSPA4, HOXC6, HLA-H, HLA-DRB3, HIF1A, GSTM2, NR3C1, CTSB, CST3, CRP, AQP3, TSPO, BTK, BDNF, BCL6, BAX, ATHS, ATD, STS, FASLG, CA1, APOH, AMY1C, AMY1B, AMY1A, AMH, ALPP, ALCAM, ADA, C4A, CA2, CR2, CD44, CP, CNN3, CLN3, CFTR, CENPB, CDKN2D, CD69, CD68, CD40LG, CAST, TNFRSF8, CD27, CD14, CD9, CD8A, CD4, CD2, CASP1, ILF2, ILF3, INSRR, RPE65, CX3CL1, CCL25, CCL22, CCL17, CCL5, SATB1, S100A9, S100A8, TRIM27, SLC17A1, RELA, PLAAT4, PVT1, PTPN2, PTN, PSMB8, PRTN3, EIF2AK2, SELE, SLC22A1, MAPK8, TARBP1, VDR, UVRAG, UBTF, TTR, TRPM2, TPMT, TNFRSF1A, CLDN5, TAP1, SLPI, TAC1, STIM1, STAT3, SSRP1, SPTBN1, SPRR2A, SPRR1B, SOAT1, MAP2K7, MAPK1, IRAK1, LTF, MSN, MSH5, MS, MPO, MMP3, MMP2, LYZ, LY9, LTBR, MYC, LPL, LEPR, LEP, LCN1, KRT6B, KDR, IRF2, IRF1, MUC4, NCL, PRKAB1, PIK3CB, PRKAA2, PRKAA1, PPBP, PPARG, PPARA, POU2F1, PIK3CG, PIK3CD, PIK3CA, NFKB1, SERPINA1, PDCD1, PAX3, PAEP, P2RX7, OXTR, NR4A2, NFKBIL1, H3P19
-
Apert Syndrome
Medlineplus
Apert syndrome is a genetic disorder characterized by skeletal abnormalities. A key feature of Apert syndrome is the premature closure of the bones of the skull (craniosynostosis). ... Individuals with Apert syndrome have syndactyly of the fingers and toes . ... Some people with Apert syndrome have abnormalities in the bones of the elbows or shoulders. ... Frequency Apert syndrome affects an estimated 1 in 65,000 to 88,000 newborns.
-
17q12 Deletion Syndrome
Medlineplus
17q12 deletion syndrome is a condition that results from the deletion of a small piece of chromosome 17 in each cell. ... The signs and symptoms of 17q12 deletion syndrome vary widely, even among affected members of the same family. ... Frequency The worldwide prevalence of 17q12 deletion syndrome is unknown, although the condition appears to be rare. One study estimated that 17q12 deletion syndrome occurs in 1 in 14,500 people in Iceland. ... The chromosome segment most commonly deleted in people with 17q12 deletion syndrome contains 15 genes. The loss of two genes in particular, HNF1B and LHX1 , is thought to underlie some of the features of 17q12 deletion syndrome.
-
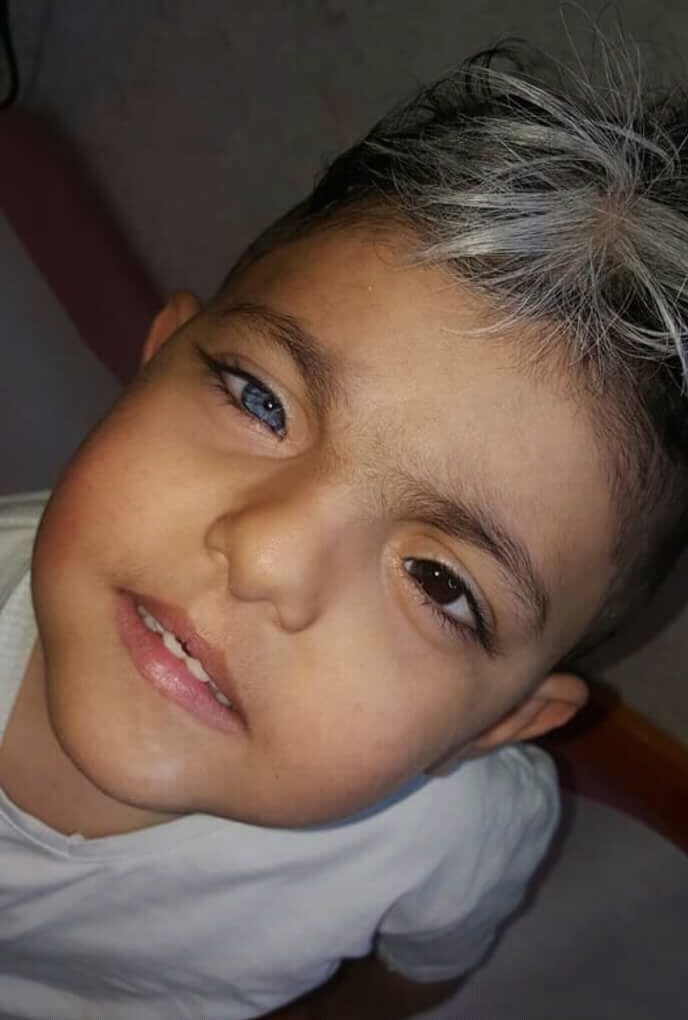
Waardenburg Syndrome
Medlineplus
Type IV (also known as Waardenburg-Shah syndrome) has signs and symptoms of both Waardenburg syndrome and Hirschsprung disease, an intestinal disorder that causes severe constipation or blockage of the intestine. Frequency Waardenburg syndrome affects an estimated 1 in 40,000 people. ... Types I and II are the most common forms of Waardenburg syndrome, while types III and IV are rare. ... Mutations in the MITF or SNAI2 gene can cause Waardenburg syndrome type II. Mutations in the SOX10 , EDN3 , or EDNRB gene can cause Waardenburg syndrome type IV. ... In some cases, the genetic cause of Waardenburg syndrome has not been identified. Learn more about the genes associated with Waardenburg syndrome EDN3 EDNRB MITF PAX3 SNAI2 SOX10 Inheritance Pattern Waardenburg syndrome is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.PAX3, MITF, SNAI2, AEBP2, WFS1, RET, EDNRB, ABO, ASS1, EYA4, FN1, GJB2, SERPINF2, SOX10, DCTN6, CISD2
-
Freeman–sheldon Syndrome
Wikipedia
"Recessive form of Freeman-Sheldon's syndrome or 'whistling face ' " . J. Med. ... "Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome". Nat. ... "[Hand deformities in Freeman-Sheldon syndrome and their surgical treatment]". ... "Anesthesia for Freeman-Sheldon syndrome using a laryngeal mask airway" . ... "[Cranio-carpo-tarsal dysplasia syndrome (Freeman-Sheldon syndrome, whistling face syndrome)]".

-
Isolated Duane Retraction Syndrome
Medlineplus
Isolated Duane retraction syndrome is a disorder of eye movement. ... Duane retraction syndrome can also occur as part of syndromes that affect other areas of the body. ... Frequency Isolated Duane retraction syndrome affects an estimated 1 in 1,000 people worldwide. ... For unknown reasons, isolated Duane syndrome affects females more often than males. ... Learn more about the gene associated with Isolated Duane retraction syndrome CHN1 Inheritance Pattern Isolated Duane retraction syndrome usually occurs in people with no history of the disorder in their family.










