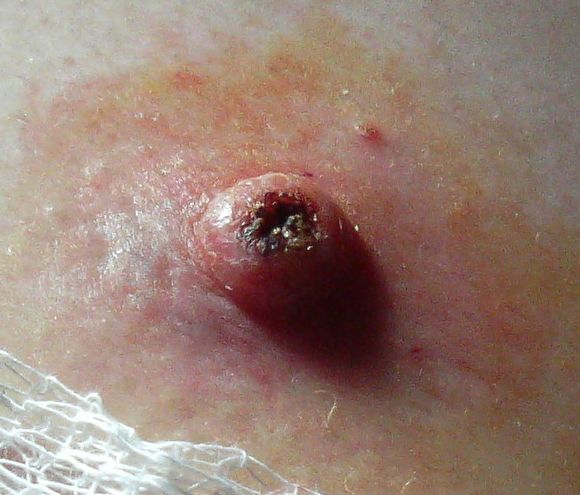
Keratoacanthoma Keratoacanthoma Specialty Dermatology , plastic surgery Types Giant keratoacanthomas Subungual keratoacanthoma Multiple keratoacanthomas (Ferguson–Smith syndrome) Keratoacanthoma centrifugum marginatum Generalized eruptive keratoacanthoma of Grzybowski Risk factors Ultraviolet radiation , immunosuppression , genetics Diagnostic method Tissue biopsy Differential diagnosis Squamous cell skin cancer Treatment Surgery ( excision , Mohs surgery ) Keratoacanthoma ( KA ) is a common low-grade (unlikely to metastasize or invade) rapidly-growing skin tumour that is believed to originate from the hair follicle ( pilosebaceous unit ) and can resemble squamous cell carcinoma . [1] [2] The defining characteristic of a keratoacanthoma is that it is dome-shaped, symmetrical, surrounded by a smooth wall of inflamed skin, and capped with keratin scales and debris. ... While some pathologists classify keratoacanthoma as a distinct entity and not a malignancy, about 6% of clinical and histological keratoacanthomas do progress to invasive and aggressive squamous cell cancers; some pathologists may label KA as "well-differentiated squamous cell carcinoma, keratoacanthoma variant", and prompt definitive surgery may be recommended. [5] [6] [7] [8] Contents 1 Classification 2 Cause 3 Diagnosis 4 Treatment 5 History 6 See also 7 References 8 External links Classification [ edit ] A person with generalized eruptive keratoacanthomas Frequently reported and reclassified over the last century, keratoacanthoma can be divided into various subtypes and despite being considered benign, their unpredictable behaviour has warranted the same attention as with squamous cell carcinoma. [1] Keratoacanthomas may be divided into the following types: [9] : 763–764 [10] : 643–646 Giant keratoacanthomas are a variant of keratoacanthoma, which may reach dimensions of several centimeters. [9] : 763 Keratoacanthoma centrifugum marginatum is a cutaneous condition, a variant of keratoacanthomas, which is characterized by multiple tumors growing in a localized area. [9] : 763 [10] : 645 Multiple keratoacanthomas (also known as "Ferguson–Smith syndrome," "Ferguson-Smith type of multiple self-healing keratoacanthomas,") is a cutaneous condition, a variant of keratoacanthomas, which is characterized by the appearance of multiple, sometimes hundreds of keratoacanthomas. [9] : 763 [10] : 644 A solitary keratoacanthoma (also known as "Subungual keratoacanthoma") is a benign, but rapidly growing, locally aggressive tumor which sometimes occur in the nail apparatus. [9] : 667,764 [10] : 644 Generalized eruptive keratoacanthoma (also known as "Generalized eruptive keratoacanthoma of Grzybowski") is a cutaneous condition, a variant of keratoacanthomas, characterized by hundreds to thousands of tiny follicular keratotic papules over the entire body. [9] : 763 [10] : 645 Treatments are not successful for many people with generalized eruptive keratoacanthoma. ... External links [ edit ] Classification D ICD - 10 : D23 ( ILDS D23.L71) (ICD10.v4 L85.8) ICD - 9-CM : 238.2 MeSH : D007636 DiseasesDB : 29383 External resources eMedicine : derm/206 Patient UK : Keratoacanthoma v t e Skin cancer of the epidermis Tumor Carcinoma BCC Forms Aberrant Cicatricial Cystic Fibroepithelioma of Pinkus Infltrative Micronodular Nodular Pigmented Polypoid Pore-like Rodent ulcer Superficial Nevoid basal cell carcinoma syndrome SCC Forms Adenoid Basaloid Clear cell Signet-ring-cell Spindle-cell Marjolin's ulcer Bowen's disease Bowenoid papulosis Erythroplasia of Queyrat Actinic keratosis Adenocarcinoma Aggressive digital papillary adenocarcinoma Extramammary Paget's disease Ungrouped Merkel cell carcinoma Microcystic adnexal carcinoma Mucinous carcinoma Primary cutaneous adenoid cystic carcinoma Verrucous carcinoma Malignant mixed tumor Benign tumors Acanthoma Forms Large cell Fissuring Clear cell Epidermolytic Melanoacanthoma Pilar sheath acanthoma Seboacanthoma Seborrheic keratosis Warty dyskeratoma Keratoacanthoma Generalized eruptive Keratoacanthoma centrifugum marginatum Multiple Solitary Wart Verruca vulgaris Verruca plana Plantar wart Periungual wart Other Epidermal nevus Syndromes Epidermal nevus syndrome Schimmelpenning syndrome Nevus comedonicus syndrome Nevus comedonicus Inflammatory linear verrucous epidermal nevus Linear verrucous epidermal nevus Pigmented hairy epidermal nevus syndrome Systematized epidermal nevus Phakomatosis pigmentokeratotica Other nevus Nevus unius lateris Patch blue nevus Unilateral palmoplantar verrucous nevus Zosteriform speckled lentiginous nevus Ungrouped Cutaneous horn
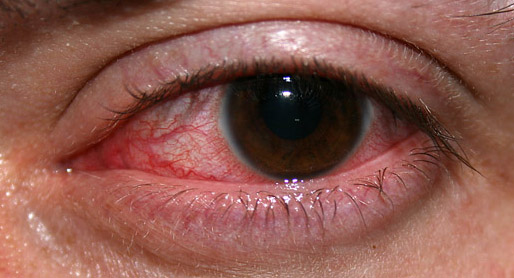
Overview Keratitis is an inflammation of the cornea — the clear, dome-shaped tissue on the front of your eye that covers the pupil and iris. Keratitis may or may not be associated with an infection. Noninfectious keratitis can be caused by a relatively minor injury, such as from wearing your contact lenses too long or getting a foreign body in the eye. Infectious keratitis can be caused by bacteria, viruses, fungi and parasites. If you have eye redness or other symptoms of keratitis, make an appointment to see an eye specialist. With prompt attention, mild to moderate cases of keratitis can usually be effectively treated without loss of vision.
Léri-Weill dyschondrosteosis is a disorder of bone growth. Affected individuals typically have shortening of the long bones in the arms and legs (mesomelia). As a result of the shortened leg bones, people with Leri-Weill dyschondrosteosis typically have short stature. Most people with the condition also have an abnormality of the wrist and forearm bones called Madelung deformity, which may cause pain and limit wrist movement. This abnormality usually appears in childhood or early adolescence. Other features of Léri-Weill dyschondrosteosis can include increased muscle mass (muscle hypertrophy); bowing of a bone in the lower leg called the tibia; a greater-than-normal angling of the elbow away from the body (increased carrying angle); and a high arched palate. Léri-Weill dyschondrosteosis occurs in both males and females, although its signs and symptoms tend to be more severe in females.
Madelung deformity (MD) is a rare congenital (present from birth) condition in which the wrist grows abnormally and part of the radius, one of the bones of the forearms, stops growing early and is short and bowed. The other forearm bone, the ulna, keeps growing and can dislocate, forming a bump. Symptoms typically develop in mid- to late-childhood or early adolescence (around 6 to 13 years of age) and usually affect both wrists. It is more commonly observed in females. Symptoms include a decreased range of motion in the wrist, pain, and a visible difference in the appearance of the wrist. In addition to the abnormal growth, there is also an abnormal palmar (Vickers’ ligament) that is thought to contribute to the deformity.
Hemizygous mutation in the EBP gene can also cause MEND syndrome (300960) in males, which shares some features with CDPX2. ... X-linked dominant CDP, also known as Conradi-Hunermann syndrome, is the most well-characterized form. ... Although Sutphen et al. (1995) suggested that their male with XXY Klinefelter syndrome was the first known case of Happle syndrome in a man, Happle (1995) pointed to reports of 3 unrelated males with this disorder, at least 2 of whom had chromosome studies showing 46,XY karyotype. ... The authors concluded that abnormal cholesterol biosynthesis is a characteristic of some syndromes with rhizomelic shortening and chondrodysplasia punctata. ... Nomenclature Kalter et al. (1989) suggested that the designation Conradi-Hunermann syndrome be reserved for the X-linked dominant form of the disorder.
X-linked dominant chondrodysplasia punctata 2 (CDPX2), also known as Conradi-Hünermann-Happle syndrome, is a rare form of skeletal dysplasia characterized by skeletal malformations, skin abnormalities, cataracts and short stature.
A syndrome characterized by bone loss Gorham's disease Other names Acro-osteolysis syndrome, Breschet-Gorham-Stout syndrome, [1] Cystic angiomatosis of bone, [1] Disappearing bone disease, Disseminated lymphangiomatosis, Disseminated osseous bone disease, Essential osteolysis, Gorham-Stout syndrome, Gorham's lymphangiomatosis, Hemangiomata with osteolysis, [1] Idiopathic massive osteolysis, Massive osteolysis, [1] Morbus-Gorham-Stout disease, Osteolysis and angiomatous nevi, [1] Skeletal lymphangiomatosis, Skeletal hemangiomatosis, Thoracic lymphangiomatosis. ... The apparent contradiction concerning the presence or absence or the number of osteoclasts, may be explained by the different phases of the syndrome." They further stated that their histopathological study provided good evidence that osteolytic changes seen in Gorham's disease are the result of hyperactive osteoclastic bone. ... S., Delling, G. "The Gorham-Stout syndrome (Gorham's massive osteolysis) A REPORT OF SIX CASES WITH HISTOPATHOLOGICAL FINDINGS." ... Plasma VEGF determination in disseminated lymphangiomatosis-Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. ... External links [ edit ] Classification D ICD - 9-CM : 733.99 } MeSH : D010015 DiseasesDB : 31515 SNOMED CT : 1515008 External resources Orphanet : 73 v t e Bone and joint disease Bone Inflammation endocrine : Osteitis fibrosa cystica Brown tumor infection : Osteomyelitis Sequestrum Involucrum Sesamoiditis Brodie abscess Periostitis Vertebral osteomyelitis Metabolic Bone density Osteoporosis Juvenile Osteopenia Osteomalacia Paget's disease of bone Hypophosphatasia Bone resorption Osteolysis Hajdu–Cheney syndrome Ainhum Gorham's disease Other Ischaemia Avascular necrosis Osteonecrosis of the jaw Complex regional pain syndrome Hypertrophic pulmonary osteoarthropathy Nonossifying fibroma Pseudarthrosis Stress fracture Fibrous dysplasia Monostotic Polyostotic Skeletal fluorosis bone cyst Aneurysmal bone cyst Hyperostosis Infantile cortical hyperostosis Osteosclerosis Melorheostosis Pycnodysostosis Joint Chondritis Relapsing polychondritis Other Tietze's syndrome Combined Osteochondritis Osteochondritis dissecans Child leg: hip Legg–Calvé–Perthes syndrome tibia Osgood–Schlatter disease Blount's disease foot Köhler disease Sever's disease spine Scheuermann's_disease arm: wrist Kienböck's disease elbow Panner disease
Treatment is anti-retroviral therapy . [5] Disseminated Histoplasmosis: During a large urban Disseminated Histoplasmosis outbreak (est. 100,000 victims) in Indianapolis from 1978–1979, manifestations included parotitis. [6] Autoimmune causes [ edit ] These are also collectively known as chronic punctate parotitis or chronic autoimmune parotitis. Sjögren's syndrome: Chronic inflammation of the salivary glands may also be an autoimmune disease known as Sjögren's syndrome . The disease most commonly appears in people aged 40–60 years, but it may affect small children. In Sjögren syndrome, the prevalence of parotitis in women versus men is approximately 9:1. The involved parotid gland is enlarged and tender at times. The cause is unknown. The syndrome is often characterized by excessive dryness in the eyes, mouth, nose, vagina, and skin. [2] Lymphoepithelial lesion of Godwin: Most frequently associated with a circumscribed tumor with the histologic features of Sjögren syndrome. ... Quiescent periods between episodes last for hours, days, or even years. [2] Recurrent parotitis of childhood: An uncommon syndrome in which recurring episodes clinically resembling mumps. ... Xerostomia occasionally occurs. The Heerfordt-Waldenstrom syndrome consists of sarcoidosis with parotid enlargement, fever, anterior uveitis, and facial nerve palsy. [2] IgG4-related sialadenitis : This term refers to IgG4-related disease (IgG4-RD) involving any of the major salivary glands, i.e. parotid or submandibular glands.
Acute flaccid myelitis : a polio-like syndrome that causes muscle weakness and paralysis. ... Others also report fever, respiratory problems and intractable vomiting. [3] Diseases associated with myelitis [ edit ] Conditions associated with myelitis include: Acute disseminated encephalomyelitis : autoimmune demyelination of the brain causing severe neurological signs and symptoms Multiple sclerosis : demyelination of the brain and spinal cord Neuromyelitis optica or Devic's disease : immune attack on optic nerve and spinal cord Sjögren's syndrome : destruction of the exocrine system of the body Systemic lupus erythematosus : a systemic autoimmune disease featuring a wide variety of neurological signs and symptoms Sarcoidosis : [4] chronic inflammatory cells form as nodules in multiple organs Atopy : an immune disorder of children manifesting as eczema or other allergic conditions. [5] It can include atopic myelitis, which causes weakness. [6] Immune-mediated myelopathies , heterogeneous group of inflammatory spinal cord disorders including autoimmune disorders with known antibodies [7] Cause [ edit ] Myelitis occurs due to various reasons such as infections. ... Studies show that NSCs that were transplanted into a demyelinating spinal cord lesion were found to regenerate oligodendrocytes and Schwann cells , and completely remyelinated axons. [18] See also [ edit ] Encephalomyelitis Myalgic encephalomyelitis – also known as chronic fatigue syndrome Transverse myelitis References [ edit ] ^ Kelly H (April 2006). ... External links [ edit ] Classification D ICD - 10 : G04 - G05 ICD - 9-CM : 323 MeSH : D009187 DiseasesDB : 29461 v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis v t e Focal lesions of the spinal cord General Myelopathy Myelitis Spinal cord compression By location Brown-Séquard syndrome Posterior cord syndrome Anterior cord syndrome Central cord syndrome Cauda equina syndrome Other Polio Demyelinating disease Transverse myelitis Tropical spastic paraparesis Epidural abscess Syringomyelia Syringobulbia Morvan's syndrome Sensory ataxia Tabes dorsalis Abadie's sign Subacute combined degeneration of spinal cord Vascular myelopathy Anterior spinal artery syndrome Foix–Alajouanine syndrome
Inflammatory diseases of the blood vessels, like giant-cell arteritis , polyarteritis nodosa, Churg-Strauss syndrome, granulomatosis with polyangiitis , and rheumatoid arthritis can cause arteritic AIONs (AAION). ... Posterior ischemic optic neuropathy is a syndrome of sudden visual loss with optic neuropathy without initial disc swelling with subsequent development of optic atrophy. ... Examination of these patients shows loss of visual acuity, temporal pallor of the optic discs, centrocecal scotomas with peripheral sparing, and subtle impairments in color vision. Behr’s syndrome is a rare autosomal recessive disorder characterized by early-onset optic atrophy, ataxia, and spasticity. Berk–Tabatznik syndrome is a condition that shows symptoms of short stature, congenital optic atrophy and brachytelephalangy. ... PMID 8071952 . ^ Genetic and Rare Diseases Information Center (GARD). "Berk-Tabatznik syndrome" . Retrieved 28 September 2013 . ^ Sadun, Alfredo A (2009).
Disseminated intravascular coagulation may occur in a significant number of patients with presentation of various degrees of thrombin generation, followed by decreased fibrinogen and increased fibrinolysis. [ citation needed ] Spontaneous tumor lysis syndrome is present in approximately 10 percent of patients with leukostasis. ... Disseminated intravascular coagulation and spontaneous tumor lysis syndrome can develop before and after chemotherapy treatment. ... Specific treatments include lysis syndrome treatment in addition to aggressive intravenous hydration with allopurinol or rasburicase to decrease serum uric acid levels. [2] Hematopoietic cell transplants are critical to correct leukostasis and leukemia. ... Such rapid and massive lysis of tissue poses a risk of complications ( tumor lysis syndrome ), but it is necessary to avoid a stroke . ... Medical Subject Headings, 2009–2009-02-13 . 1995-06-01. v t e Diseases of monocytes and granulocytes Monocytes and macrophages ↑ -cytosis : Monocytosis Histiocytosis Chronic granulomatous disease ↓ -penia : Monocytopenia Granulocytes ↑ -cytosis : granulocytosis Neutrophilia Eosinophilia / Hypereosinophilic syndrome Basophilia Bandemia ↓ -penia : Granulocytopenia/agranulocytosis ( Neutropenia / Severe congenital neutropenia / Cyclic neutropenia Eosinopenia Basopenia ) Disorder of phagocytosis Chemotaxis and degranulation Leukocyte adhesion deficiency LAD1 LAD2 Chédiak–Higashi syndrome Neutrophil-specific granule deficiency Respiratory burst Chronic granulomatous disease Neutrophil immunodeficiency syndrome Myeloperoxidase deficiency v t e Lymphatic disease : organ and vessel diseases Thymus Abscess Hyperplasia Hypoplasia DiGeorge syndrome Ectopic thymus Thymoma Thymic carcinoma Spleen Asplenia Asplenia with cardiovascular anomalies Accessory spleen Polysplenia Wandering spleen Splenomegaly Banti's syndrome Splenic infarction Splenic tumor Lymph node Lymphadenopathy Generalized lymphadenopathy Castleman's disease Intranodal palisaded myofibroblastoma Kikuchi disease Tonsils see Template:Respiratory pathology Lymphatic vessels Lymphangitis Lymphangiectasia Lymphedema Primary lymphedema Congenital lymphedema Lymphedema praecox Lymphedema tarda Lymphedema–distichiasis syndrome Milroy's disease Secondary lymphedema Bullous lymphedema Factitial lymphedema Postinflammatory lymphedema Postmastectomy lymphangiosarcoma Waldmann disease
Synonyms for ORS, many historical, include bromidrosiphobia, [2] olfactory phobic syndrome, [2] chronic olfactory paranoid syndrome, [2] autodysomophobia, [2] delusions of bromosis, [5] hallucinations of smell [5] and olfactory delusional syndrome. [ citation needed ] By definition, the many terms which have been suggested in the dental literature to refer to subjective halitosis complaints (i.e. where a person complains of halitosis yet no odor is detectable clinically) can also be considered under the umbrella of ORS. ... "Delusions of body malodour: the olfactory reference syndrome." (PDF) . In Brewer WJ, Castle D, Pantelis C (eds.). ... PMID 9051972 . ^ a b c Phillips, KA; Menard, W (Jul–Aug 2011). "Olfactory reference syndrome: demographic and clinical features of imagined body odor" . ... "How to help patients with olfactory reference syndrome" (PDF) . Current Psychiatry . 6 (3). ... "Trimethylaminuria (fish malodour syndrome): a "benign" genetic condition with major psychosocial sequelae".
Overview An ischemic stroke occurs when the blood supply to part of the brain is interrupted or reduced, preventing brain tissue from getting oxygen and nutrients. Brain cells begin to die in minutes. A stroke is a medical emergency, and prompt treatment is crucial. Early action can reduce brain damage and other complications. The good news is that many fewer Americans die of stroke now than in the past. Effective treatments can also help prevent disability from stroke. Symptoms If you or someone you're with may be having a stroke, pay particular attention to the time the symptoms began. Some treatment options are most effective when given soon after a stroke begins.
The 24-hour limit divides stroke from transient ischemic attack , which is a related syndrome of stroke symptoms that resolve completely within 24 hours. [2] With the availability of treatments that can reduce stroke severity when given early, many now prefer alternative terminology, such as brain attack and acute ischemic cerebrovascular syndrome (modeled after heart attack and acute coronary syndrome , respectively), to reflect the urgency of stroke symptoms and the need to act swiftly. [20] Ischemic Main articles: Cerebral infarction and Brain ischemia In an ischemic stroke, blood supply to part of the brain is decreased, leading to dysfunction of the brain tissue in that area. ... In paradoxical embolism , a deep vein thrombosis embolizes through an atrial or ventricular septal defect in the heart into the brain. [45] Causes of stroke related to the heart can be distinguished between high and low-risk: [46] High risk: atrial fibrillation and paroxysmal atrial fibrillation , rheumatic disease of the mitral or aortic valve disease, artificial heart valves , known cardiac thrombus of the atrium or ventricle, sick sinus syndrome , sustained atrial flutter , recent myocardial infarction, chronic myocardial infarction together with ejection fraction <28 percent, symptomatic congestive heart failure with ejection fraction <30 percent, dilated cardiomyopathy , Libman-Sacks endocarditis , Marantic endocarditis , infective endocarditis , papillary fibroelastoma , left atrial myxoma and coronary artery bypass graft (CABG) surgery.
Description Testicular microlithiasis, the deposition of calcium phosphate microliths within the seminiferous tubules, has a population prevalence of 0.6 to 9% (Kim et al., 2003). Middleton et al. (2002) found that it was associated with a majority of primary testicular malignancies. Miller and Sidhu (2002) found that it was present in 1% of male idiopathic infertility cases. Clinical Features Coffey et al. (2007) analyzed the frequency of testicular microlithiasis (TM) in 169 patients with testicular germ cell tumor (TGCT; 273300), 58 relatives, and 101 controls, and found that TM was more frequent in cases than controls (age-adjusted p less than 0.0001) and in unaffected male relatives than controls (age-adjusted p = 0.02). TGCT cases and matched relative pairs showed greater concordance for TM than would be expected by chance alone (p = 0.05).