Testicular histology in both brothers revealed Sertoli cell-only syndrome, with complete lack of sperm and extensive sclerosis and tubular atrophy. ... Molecular Genetics In 2 Estonian brothers with nonobstructive azoospermia and Sertoli cell-only syndrome, Kasak et al. (2018) performed whole-exome sequencing and identified compound heterozygosity for a 1-bp duplication (609644.0003) and a splice site mutation (609644.0004) in the FANCM gene. ... Noting that testicular histology was unavailable for the latter 2 patients, the authors stated that the association between loss-of-function variants in FANCM and Sertoli cell-only syndrome remained to be verified in future patients. ... INHERITANCE - Autosomal recessive GENITOURINARY External Genitalia (Male) - Small testes Internal Genitalia (Male) - Azoospermia, nonobstructive - Oligoasthenospermia (in some patients) - Sertoli cell-only syndrome ENDOCRINE FEATURES - Elevated follicle-stimulating hormone (FSH) - Elevated luteinizing hormone (LH) - Low testosterone MOLECULAR BASIS - Caused by mutation in the FANCM gene (FANCM, 609644.0003 ) ▲ Close
Clinical Features Bouwes Bavinck et al. (1987) reported a mother and son with an apparently previously undescribed syndrome involving mild mental retardation, microcephaly, short stature, eye defects, hypoplastic and posteriorly rotated low-set ears with overfolded helices, and mild facial dysmorphism, including widow's peak, narrow bifrontal diameter, midface hypoplasia, depressed nasal bridge, thickened alae nasi, and long flat philtrum. ... Although the disorder was similar to the autosomal dominant or X-linked dominant MCA/MR syndrome characterized by microcephaly, eye anomalies, short stature, and mental deficiency reported by Bouwes Bavinck et al. (1987), and to the trichorhinophalangeal syndromes (see 190350), Kondoh et al. (2001) concluded that it was distinct. ... The 2 mothers of these boys also showed features of the syndrome, including short stature, dysmorphic facial features, and mild cortical cataract, but were significantly less severely affected than their children.
Cytogenetics Laryngeal web associated with deletions and microdeletions of 22q11.2, with or without signs of the velocardiofacial syndrome (192430) or DiGeorge syndrome (188400), was noted by Gay et al. (1981), Driscoll et al. (1992), Lindsay et al. (1995), and Fokstuen et al. (1997). ... One patient showed a 'classic' 22q11.2 deletion phenotype with clinical overlap with DiGeorge syndrome and velocardiofacial syndrome.
Baritosis is an extremely rare, benign form of pneumoconiosis that causes little or no overgrowth, hardening, and/or scarring of the tissue in the lung (fibrosis). Pneumoconiosis is caused by accumulation of inhaled particles and involves a reaction of tissue in the lung. In the case of baritosis, the inhaled particles are made up of barium sulfate and is well described in workers who crush and grind compounds containing barium, a mineral found in paints, paper, ceramics, glass, rubber, electronic components, and in drilling muds in oil and gas exploration. Baritosis is typically characterized by a mixture of very fine punctate and annular (ring-like) lesions and some slightly larger nodular lesions in the lung. The condition generally appears 1 to 2 years after exposure, does not affect the function of the lung, and appears to go away without treatment after exposure stops.
With some other medical conditions, the root cause for a large percentage of all cases have not been established—for example, focal segmental glomerulosclerosis or ankylosing spondylitis ; the majority of these cases are deemed idiopathic. [2] Contents 1 Medical advances and this term 2 Usage of synonyms 3 Syndrome without a name 4 See also 5 References 6 External links Medical advances and this term [ edit ] Advances in medical science improve the understanding of causes of diseases and the classification of diseases; thus, regarding any particular condition or disease, as more root causes are discovered and as events that seemed spontaneous have their origins revealed, the percentage of cases designated as idiopathic decreases. ... Some congenital conditions are idiopathic, and sometimes the word congenital is used synonymously with idiopathic ; but careful usage prefers to reserve the word congenital for conditions to which the literal sense of the word applies (that is, those whose pathophysiology has existed since the neonatal period). Syndrome without a name [ edit ] The term syndrome(s) without a name (SWAN) is used "when a child or young adult is believed to have a genetic condition and testing has failed to identify its genetic cause". ... Genetic Alliance UK. 22 March 2017 . Retrieved 16 April 2019 . ^ "Syndromes without a name (SWAN)" . Raising Children Network (Australia). 10 January 2017 .
Contents 1 Symptoms and diagnosis 2 Treatment 3 Research 4 See also 5 References Symptoms and diagnosis [ edit ] Chronic idiopathic constipation is similar to constipation-predominant irritable bowel syndrome (IBS-C); however, people with CIC do not have other symptoms of IBS, such as abdominal pain. [1] Diagnosing CIC can be difficult as other syndromes must be ruled out as there is no physiological cause for CIC. ... To be considered functional constipation, symptoms must be present at least a fourth of the time. [1] Possible causes are: Anismus Descending perineum syndrome Other inability or unwillingness to control the external anal sphincter , which normally is under voluntary control A poor diet An unwillingness to defecate Nervous reactions, including prolonged and/or chronic stress and anxiety, that close the internal anal sphincter , a muscle that is not under voluntary control Deeper psychosomatic disorders which sometimes affect digestion and the absorption of water in the colon There is also possibility of presentation with other comorbid symptoms such as headache, especially in children. [2] Treatment [ edit ] Treatment options appear similar and include prucalopride , lubiprostone , linaclotide , tegaserod , velusetrag , elobixibat , bisacodyl , sodium picosulphate , [3] and most recently, plecanatide . ... "Efficacy of Prebiotics, Probiotics, and Synbiotics in Irritable Bowel Syndrome and Chronic Idiopathic Constipation: Systematic Review and Meta-analysis".
This condition may be a congenital defect associated with underlying abnormalities of the pectoral muscle (as in Poland's syndrome [2] ), related to trauma (typically surgery or radiotherapy ) or it may be a more subjective aesthetic description. ... Contents 1 Causes 2 Treatment 3 See also 4 References 5 External links Causes [ edit ] Micromastia can be congenital or acquired disorder and may be unilateral or bilateral. [3] Congenital causes include ulnar–mammary syndrome (caused by mutations in the TBX3 gene ), Poland syndrome , Turner syndrome , and congenital adrenal hyperplasia . [3] There is also a case report of familial hypoplasia of the nipples and athelia associated with mammary hypoplasia that was described in a father and his daughters. [3] Acquired causes of micromastia include irradiation in infancy and childhood and surgical removal of prepubertal breast bud . [3] Treatment [ edit ] The procedure to remedy micromastia is breast enlargement , most commonly augmentation mammoplasty using breast implants .
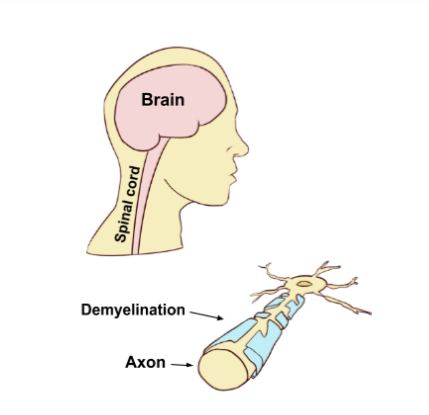
Cite journal requires |journal= ( help ) External links [ edit ] Classification D MeSH : D020278 v t e Multiple sclerosis and other demyelinating diseases of the central nervous system Signs and symptoms Ataxia Depression Diplopia Dysarthria Dysphagia Fatigue Incontinence Nystagmus Optic neuritis Pain Uhthoff's phenomenon Investigations and diagnosis Multiple sclerosis diagnosis McDonald criteria Poser criteria Clinical Clinically isolated syndrome Expanded Disability Status Scale Serological and CSF Oligoclonal bands Radiological Radiologically isolated syndrome Lesional demyelinations of the central nervous system Dawson's fingers Approved [ by whom? ] treatment Management of multiple sclerosis Alemtuzumab Cladribine Dimethyl fumarate Fingolimod Glatiramer acetate Interferon beta-1a Interferon beta-1b Mitoxantrone Natalizumab Ocrelizumab Ozanimod Siponimod Teriflunomide Other treatments Former Daclizumab Multiple sclerosis research Demyleinating diseases Autoimmune Multiple sclerosis Neuromyelitis optica Diffuse myelinoclastic sclerosis Inflammatory Acute disseminated encephalomyelitis MOG antibody disease Balo concentric sclerosis Marburg acute multiple sclerosis Neuromyelitis optica Diffuse myelinoclastic sclerosis Tumefactive multiple sclerosis Experimental autoimmune encephalomyelitis Hereditary Adrenoleukodystrophy Alexander disease Canavan disease Krabbe disease Metachromatic leukodystrophy Pelizaeus–Merzbacher disease Leukoencephalopathy with vanishing white matter Megalencephalic leukoencephalopathy with subcortical cysts CAMFAK syndrome Other Central pontine myelinolysis Marchiafava–Bignami disease Mitochondrial DNA depletion syndrome Other List of multiple sclerosis organizations List of people with multiple sclerosis Multiple sclerosis drug pipeline Pathophysiology This article about a medical condition affecting the nervous system is a stub .
Brain imaging of 1 of the patients did not show evidence of Leigh syndrome. Three sibs in a second family with isolated complex I deficiency presented in infancy with seizures and developmental regression. ... Brain imaging in these patients showed progressive brain atrophy and changes in the basal ganglia consistent with Leigh syndrome. One died at age 10 year, 1 was in a vegetative state by age 3 and was still alive at age 32, and the third died at 19 months of age. ... Two sibs from another family with MC1DN7 manifest as early-onset seizures and Leigh syndrome were subsequently found to be homozygous for the c.IVS2+1delGTAA mutation by direct sequencing of the gene in 10 additional families. ... INHERITANCE - Autosomal recessive GROWTH Other - Failure to thrive HEAD & NECK Head - Microcephaly Eyes - Nystagmus - Optic atrophy CARDIOVASCULAR Heart - Hypertrophic cardiomyopathy (in some patients) MUSCLE, SOFT TISSUES - Hypotonia - Skeletal muscle biopsy shows variation in fiber size - Atrophic fibers - Subsarcolemmal accumulation of mitochondria NEUROLOGIC Central Nervous System - Encephalopathy - Impaired psychomotor development - Developmental regression - Spasticity - Seizures, early-onset (in some patients) - Brain atrophy - Abnormalities consistent with Leigh syndrome seen on brain imaging (in some patients) METABOLIC FEATURES - Lactic acidosis LABORATORY ABNORMALITIES - Mitochondrial respiratory complex I deficiency in various tissues - Increased lactate:pyruvate ratio MISCELLANEOUS - Onset in infancy - Variable features - Early death may occur MOLECULAR BASIS - Caused by mutation in the NADH-ubiquinone oxidoreductase flavoprotein 2 gene (NDUFV2, 600532.0002 ) ▲ Close
Because the condition causes both vaginal and urinary symptoms, doctors use the term "genitourinary syndrome of menopause (GSM)" to describe vaginal atrophy and its accompanying symptoms. Simple, effective treatments for genitourinary syndrome of menopause (GSM) are available. ... Sexual activity, with or without a partner, increases blood flow and makes your vaginal tissues more elastic. Complications Genitourinary syndrome of menopause increases your risk of: Vaginal infections. ... Diagnosis Diagnosis of genitourinary syndrome of menopause (GSM) may involve: Pelvic exam, during which your doctor feels your pelvic organs and visually examines your external genitalia, vagina and cervix. ... Treatment To treat genitourinary syndrome of menopause, your doctor may first recommend over-the-counter treatment options, including: Vaginal moisturizers.
Syndrome that causes episodes of increased activity of the sympathetic nervous system Paroxysmal sympathetic hyperactivity Specialty Neurology Paroxysmal sympathetic hyperactivity ( PSH ) is a syndrome that causes episodes of increased activity of the sympathetic nervous system. Hyperactivity of the sympathetic nervous system can manifest as increased heart rate, increased respiration, increased blood pressure, diaphoresis , and hyperthermia . [1] Previously, this syndrome has been identified as general dysautonomia but now is considered a specific form of it. ... The exact pathways or causes for the development of the syndrome are not known. Traumatic brain injury , hypoxia, [4] stroke , anti-NMDA receptor encephalitis (although further associations are being explored), [5] injury of the spinal cord , [1] and many other forms of brain injury can cause onset of PSH. ... It causes decreases in respiration, but it can be very dangerous for the liver. [3] Again, these treatments are seen case by case and treat symptoms well. They do not treat the syndrome as a whole or preventatively. Efficacy varies patient to patient, as symptoms do. ... Curr Neurol Neurosci Rep . 13 (370). doi : 10.1007/s11910-013-0370-3 . ^ Renal Sympathetic Denervation, From Wikipedia, the free encyclopedia12/7/2014 External links [ edit ] Classification D ICD - 10 : G90 ICD - 9-CM : 337.9 v t e Diseases of the autonomic nervous system General Dysautonomia Autonomic dysreflexia Autonomic neuropathy Pure autonomic failure Hereditary Hereditary sensory and autonomic neuropathy Familial dysautonomia Congenital insensitivity to pain with anhidrosis Orthostatic intolerance Orthostatic hypotension Postural orthostatic tachycardia syndrome Other Horner's syndrome Multiple system atrophy
Play media Video explanation It is the second most common cause of nephrotic syndrome in adults, with focal segmental glomerulosclerosis (FSGS) recently becoming the most common. [1] Contents 1 Signs and symptoms 2 Causes 2.1 Primary/idiopathic 2.2 Secondary 3 Pathogenesis 4 Morphology 5 Treatment 5.1 Immunosuppressive therapy options 6 Prognosis 7 Terminology 8 References 9 External links Signs and symptoms [ edit ] Most people will present as nephrotic syndrome , with the triad of albuminuria , edema and low serum albumin (with or without kidney failure ). ... Membranous nephropathy in particular is known to increase this risk more than other causes of nephrotic syndrome though the reason for this is not yet clear. ... The twin aims of treating membranous nephropathy are first to induce a remission of the nephrotic syndrome and second to prevent the development of end-stage kidney failure. ... "Immunosuppressive treatment for idiopathic membranous nephropathy in adults with nephrotic syndrome" . The Cochrane Database of Systematic Reviews (10): CD004293. doi : 10.1002/14651858.CD004293.pub3 . ... "Corticosteroids and ciclosporin A in idiopathic membranous nephropathy: higher remission rates of nephrotic syndrome and less adverse reactions than after traditional treatment with cytotoxic drugs".
Neck pain Decreased pupil size with drooping of the upper eyelid ( Horner syndrome ) Pulsatile tinnitus Ischemic signs and symptoms Temporary vision loss Ischemic stroke Causes [ edit ] dissection in ultrasound The causes of internal carotid artery dissection can be broadly categorised into two classes: spontaneous or traumatic. Spontaneous [ edit ] Once considered uncommon, spontaneous carotid artery dissection is an increasingly recognised cause of stroke that preferentially affects the middle-aged. [5] The incidence of spontaneous carotid artery dissection is low, and incidence rates for internal carotid artery dissection have been reported to be 2.6 to 2.9 per 100,000. [6] Observational studies and case reports published since the early 1980s show that patients with spontaneous internal carotid artery dissection may also have a history of stroke in their family and/or hereditary connective tissue disorders, such as Marfan syndrome , Ehlers-Danlos syndrome , autosomal dominant polycystic kidney disease , pseudoxanthoma elasticum , fibromuscular dysplasia , and osteogenesis imperfecta type I. [7] IgG4-related disease involving the carotid artery has also been observed as a cause. [8] However, although an association with connective tissue disorders does exist, most people with spontaneous arterial dissections do not have associated connective tissue disorders. Also, the reports on the prevalence of hereditary connective tissue diseases in people with spontaneous dissections are highly variable, ranging from 0% to 0.6% in one study to 5% to 18% in another study. [7] Internal carotid artery dissection can also be associated with an elongated styloid process (known as Eagle syndrome when the elongated styloid process causes symptoms). [9] [10] Traumatic [ edit ] Carotid artery dissection is thought to be more commonly caused by severe violent trauma to the head and/or neck. ... "Internal carotid dissection caused by an elongated styloid process (Eagle syndrome)" . BMJ Case Reports . 2013 : bcr2013009878. doi : 10.1136/bcr-2013-009878 . ... External links [ edit ] Classification D ICD - 10 : I72.0 ICD - 9-CM : 443.21 DiseasesDB : 2145 External resources eMedicine : emerg/82 v t e Cardiovascular disease (vessels) Arteries , arterioles and capillaries Inflammation Arteritis Aortitis Buerger's disease Peripheral artery disease Arteriosclerosis Atherosclerosis Foam cell Fatty streak Atheroma Intermittent claudication Critical limb ischemia Monckeberg's arteriosclerosis Arteriolosclerosis Hyaline Hyperplastic Cholesterol LDL Oxycholesterol Trans fat Stenosis Carotid artery stenosis Renal artery stenosis Other Aortoiliac occlusive disease Degos disease Erythromelalgia Fibromuscular dysplasia Raynaud's phenomenon Aneurysm / dissection / pseudoaneurysm torso : Aortic aneurysm Abdominal aortic aneurysm Thoracic aortic aneurysm Aneurysm of sinus of Valsalva Aortic dissection Aortic rupture Coronary artery aneurysm head / neck Intracranial aneurysm Intracranial berry aneurysm Carotid artery dissection Vertebral artery dissection Familial aortic dissection Vascular malformation Arteriovenous fistula Arteriovenous malformation Telangiectasia Hereditary hemorrhagic telangiectasia Vascular nevus Cherry hemangioma Halo nevus Spider angioma Veins Inflammation Phlebitis Venous thrombosis / Thrombophlebitis primarily lower limb Deep vein thrombosis abdomen Hepatic veno-occlusive disease Budd–Chiari syndrome May–Thurner syndrome Portal vein thrombosis Renal vein thrombosis upper limb / torso Mondor's disease Paget–Schroetter disease head Cerebral venous sinus thrombosis Post-thrombotic syndrome Varicose veins Gastric varices Portacaval anastomosis Caput medusae Esophageal varices Hemorrhoid Varicocele Other Chronic venous insufficiency Chronic cerebrospinal venous insufficiency Superior vena cava syndrome Inferior vena cava syndrome Venous ulcer Arteries or veins Angiopathy Macroangiopathy Microangiopathy Embolism Pulmonary embolism Cholesterol embolism Paradoxical embolism Thrombosis Vasculitis Blood pressure Hypertension Hypertensive heart disease Hypertensive emergency Hypertensive nephropathy Essential hypertension Secondary hypertension Renovascular hypertension Benign hypertension Pulmonary hypertension Systolic hypertension White coat hypertension Hypotension Orthostatic hypotension
TdP can be acquired by inheritance of a congenital long QT syndrome, or more commonly from the ingestion of a pharmacologic drug. ... "Incidence and clinical features of the quinidine-associated long QT syndrome: Implications for care". American Heart Journal . 111 (6): 1088–93. doi : 10.1016/0002-8703(86)90010-4 . ... "Mechanisms, risk factors, and management of acquired long QT syndrome: a comprehensive review" . Scientific World Journal . 2012 : 212178. doi : 10.1100/2012/212178 . ... CS1 maint: multiple names: authors list ( link ) ^ a b Khan IA (2002). "Long QT syndrome: diagnosis and management". American Heart Journal . 143 (1): 7–14. doi : 10.1067/mhj.2002.120295 . ... "Mechanisms, risk factors, and management of acquired long QT syndrome: a comprehensive review" . Scientific World Journal . 2012 : 212178. doi : 10.1100/2012/212178 .
The results suggest that perhaps in certain tasks (i.e., receptive labeling), echolalia should not be eliminated, but taken advantage of as it may facilitate acquisition and generalization for autistic children. [15] Tourette syndrome [ edit ] Echolalia and echopraxia are distinguishing tics of Tourette syndrome (TS); [1] the echolalic repetitions of individuals with TS are mainly echoes from within their own "tic repertoire". [1] Evidence points to a healthy mirror neuron system (MNS) but "inadequate imitation-control mechanism, which make them vulnerable to interferences". ... "The pathophysiology of echopraxia/echolalia: relevance to Gilles de la Tourette syndrome". Mov. Disord . 27 (10): 1222–9. doi : 10.1002/mds.25103 . ... PMID 7844096 . ^ Bashe, P. R. The OASIS Guide to Asperger Syndrome; Advice, Support, Insight, and Inspiration . ... Classification D ICD - 10 : R48.8 ICD - 9-CM : 784.69 MeSH : D004454 v t e Tourette syndrome Main Causes and origins History Societal and cultural aspects Management Terms Coprolalia Copropraxia Echolalia Echophenomenon Echopraxia Palilalia Palipraxia PANDAS Premonitory urge Sensory phenomena Tic Tic disorder Tourettism People Jean-Martin Charcot Donald J. ... Shapiro Organizations Tourette Association of America Tourette Canada Tourettes Action Yale Child Study Center Media Front of the Class Hichki I Have Tourette's but Tourette's Doesn't Have Me John's Not Mad " Le Petit Tourette " Maze Motherless Brooklyn Quit It The Secret Life of Lele Pons The Tic Code Tic Talk: Living with Tourette Syndrome