Contents 1 Comparison with night eating syndrome 2 Signs and symptoms 3 Diagnosis 3.1 Criteria 4 Treatment 5 History 6 See also 7 References 8 External links Comparison with night eating syndrome [ edit ] NSRED is closely related to night eating syndrome (NES) except for the fact that those suffering from NES are completely awake and aware of their eating and bingeing at night while those suffering from NSRED are sleeping and unaware of what they are doing. ... Nevertheless, Augur also said, "27 percent of subjects had RLS ( restless legs syndrome , a condition known to respond to this medication), and number and duration of waking episodes related to eating behaviors were unchanged." [3] Encouraged by the positive response verified in the above-mentioned trial treatment, doctors and psychiatrists conducted a more recent study described by Auger as "efficacy of topiramate [an antiepileptic drug associated with weight loss] in 17 consecutive patients with NSRED." ... History [ edit ] The first case of NSRED was reported in 1955, but over the next 36 years, only nine more reports were made of this syndrome. Seven of these reports were single-case studies and the other two instances were seen during objective sleep studies, all done by psychiatrists and doctors. [4] Schenck and Mahowald were the first to a major study on this disorder. ... Furthermore, unlike the patients in Stunkard's series, none of our patients had problematic eating in the evening between dinner and bedtime; sleep onset insomnia was not present; and sleep latency was usually brief, apart from several patients with RLS." [4] After realizing what was wrong with them, many of Schenck and Mahowald's patients with NSRED restricted their day eating and over exercised. [4] This table summary identifies the first initial findings concerning NSRED, and it shows how NSRED is a random malady that affects many different types of people in individual ways. See also [ edit ] Night eating syndrome References [ edit ] ^ Inoue, Y. (2015). ... "Sleep-Related Eating Disorder and Night Eating Syndrome: Sleep Disorders, Eating Disorders, Or both?"
It is associated with the group of skin and nerve syndromes called the ectodermal dysplasias . Anodontia is usually part of a syndrome and seldom occurs as an isolated entity. ... Anodontia alone will not have an affect on any other body part besides teeth being missing. [4] Associated syndromes [ edit ] Hypodontia and anodontia are frequently associated with a multitude of genetic disorders and syndromes, approximately 70. ... Three syndromes which classically have signs of anodontia are oculomandibulodyscephaly, mesoectodermal dysplasia and ectodermal dysplasia. ... References [ edit ] ^ Vahid-Dastjerdi E, Borzabadi-Farahani A, Mahdian M, Amini N (2010). "Non-syndromic hypodontia in an Iranian orthodontic population".
Can be isolated or associated with syndromes such as ectodermal dysplasia and Downs syndrome. ... "Hypodontia in patients with Down's syndrome". Collegium Antropologicum . 22 Suppl: 69–72. ... "Prevalence and characteristics of non-syndromic hypodontia among Turkish orthodontic patient population" . ... "Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis". ... "Mutations in the MSX1 gene in Turkish children with non-syndromic tooth agenesis and other dental anomalies" .
Epidemiology The prevalence is unknown but it is extremely rare and usually only occurs as part of an associated syndrome such as X-linked hypohidrotic ectodermal dysplasia (X-linked HED; see this term).
Clinical Features Cramer (1947) and Ribble (1931) observed affected sisters, and Warr (1938) described parental consanguinity. The primary dentition was not affected and no associated abnormalities were noted. Gorlin (1979) knew of at least 8 reports of complete absence of the permanent dentition with the entire primary dentition present and erupted at a normal time. Gorlin (1979) and Gorlin et al. (1980) presented evidence of autosomal recessive inheritance, including multiple affected sibs and consanguineous parents. On the basis of 2 families in which both parents had pegged or missing maxillary lateral incisors (150400), Witkop (1987) concluded that agenesis of the permanent teeth can be an expression of the homozygous state of the mutated gene.
Anodontia is a dental condition characterized by complete absence of teeth. The primary (baby) or permanent (adult) teeth may be involved. Anodontia is extremely rare when present in a pure form (without associated abnormalities). In most cases, the phenomenon is associated with a group of conditions called the ectodermal dysplasias . In these cases, abnormalities are also noted in the hair, nails, and sweat glands. Anodontia is an autosomal recessive condition. A specific gene has not yet been identified.
No evidence of a myelodysplastic syndrome no significant dysplasia no cytogenetic abnormalities suggestive of myelodysplasia Treatment [ edit ] Indications [ edit ] Not all those affected will require treatment at presentation. [10] [11] [12] People are usually split up into low and high risk for bleeding/blood clotting groups (based on their age, their medical history, their blood counts and their lifestyles), low risk individuals are usually treated with aspirin , whereas those at high risk are given hydroxycarbamide and/or other treatments that reduce platelet count (such as interferon-α and anagrelide ). [1] [10] [11] [12] Agents [ edit ] Hydroxycarbamide , interferon-α and anagrelide can lower the platelet count. ... External links [ edit ] Classification D ICD - 10 : D75.2 , D47.3 ICD - 9-CM : 238.71 ICD-O : M9962/3 OMIM : 187950 MeSH : D013920 DiseasesDB : 4522 External resources MedlinePlus : 000543 eMedicine : med/2266 v t e Disorders of bleeding and clotting Coagulation · coagulopathy · Bleeding diathesis Clotting By cause Clotting factors Antithrombin III deficiency Protein C deficiency Activated protein C resistance Protein S deficiency Factor V Leiden Prothrombin G20210A Platelets Sticky platelet syndrome Thrombocytosis Essential thrombocythemia DIC Purpura fulminans Antiphospholipid syndrome Clots Thrombophilia Thrombus Thrombosis Virchow's triad Trousseau sign of malignancy By site Deep vein thrombosis Bancroft's sign Homans sign Lisker's sign Louvel's sign Lowenberg's sign Peabody's sign Pratt's sign Rose's sign Pulmonary embolism Renal vein thrombosis Bleeding By cause Thrombocytopenia Thrombocytopenic purpura : ITP Evans syndrome TM TTP Upshaw–Schulman syndrome Heparin-induced thrombocytopenia May–Hegglin anomaly Platelet function adhesion Bernard–Soulier syndrome aggregation Glanzmann's thrombasthenia platelet storage pool deficiency Hermansky–Pudlak syndrome Gray platelet syndrome Clotting factor Hemophilia A/VIII B/IX C/XI von Willebrand disease Hypoprothrombinemia/II Factor VII deficiency Factor X deficiency Factor XII deficiency Factor XIII deficiency Dysfibrinogenemia Congenital afibrinogenemia Signs and symptoms Bleeding Bruise Hematoma Petechia Purpura Nonthrombocytopenic purpura By site head Epistaxis Hemoptysis Intracranial hemorrhage Hyphema Subconjunctival hemorrhage torso Hemothorax Hemopericardium Pulmonary hematoma abdomen Gastrointestinal bleeding Hemobilia Hemoperitoneum Hematocele Hematosalpinx joint Hemarthrosis v t e Myeloid -related hematological malignancy CFU-GM / and other granulocytes CFU-GM Myelocyte AML : Acute myeloblastic leukemia M0 M1 M2 APL/M3 MP Chronic neutrophilic leukemia Monocyte AML AMoL/M5 Myeloid dendritic cell leukemia CML Philadelphia chromosome Accelerated phase chronic myelogenous leukemia Myelomonocyte AML M4 MD-MP Juvenile myelomonocytic leukemia Chronic myelomonocytic leukemia Other Histiocytosis CFU-Baso AML Acute basophilic CFU-Eos AML Acute eosinophilic MP Chronic eosinophilic leukemia / Hypereosinophilic syndrome MEP CFU-Meg MP Essential thrombocytosis Acute megakaryoblastic leukemia CFU-E AML Erythroleukemia/M6 MP Polycythemia vera MD Refractory anemia Refractory anemia with excess of blasts Chromosome 5q deletion syndrome Sideroblastic anemia Paroxysmal nocturnal hemoglobinuria Refractory cytopenia with multilineage dysplasia CFU-Mast Mastocytoma Mast cell leukemia Mast cell sarcoma Systemic mastocytosis Mastocytosis : Diffuse cutaneous mastocytosis Erythrodermic mastocytosis Adult type of generalized eruption of cutaneous mastocytosis Urticaria pigmentosa Mast cell sarcoma Solitary mastocytoma Systemic mastocytosis Xanthelasmoidal mastocytosis Multiple/unknown AML Acute panmyelosis with myelofibrosis Myeloid sarcoma MP Myelofibrosis Acute biphenotypic leukaemia
A number sign (#) is used with this entry because thrombocythemia-1 (THCYT1) is caused by heterozygous mutation in the thrombopoietin gene (THPO; 600044) on chromosome 3q27. Description Thrombocythemia, or thrombocytosis, is a myeloproliferative disorder characterized by excessive platelet production resulting in increased numbers of circulating platelets. Thrombocythemia can be associated with thrombotic or hemorrhagic episodes and occasional leukemic transformation (summary by Wiestner et al., 1998). Genetic Heterogeneity of Thrombocythemia THCYT2 (601977) is caused by germline or somatic mutation in the THPO receptor gene (MPL; 159530) on chromosome 1p34; THCYT3 (614521) is caused by germline or somatic mutation in the JAK2 gene (147796) on chromosome 9p; and a possible X-linked form (THCYTX; 300331) has been reported. Somatic mutations in the TET2 (612839), ASXL1 (612990), SH2B3 (605093), and SF3B1 (605590) genes have also been found in cases of essential thrombocythemia.
Essential thrombocythemia belongs to a group of diseases called myeloproliferative neoplasms , which cause the bone marrow to make too many platelets, white blood cells and/or red blood cells. In essential thrombocythemia, the body produces too many platelets. The signs and symptoms vary from person to person, but most people with essential thrombocythemia do not have any symptoms when the platelet cell count first increases. Signs and symptoms that develop as the disease progresses include: increased production of megakaryocytes (a type of cell in the bone marrow that is responsible for making platelets); enlargement of the spleen ( splenomegaly ); and bleeding in several parts of the body and/or clotting episodes such as strokes, pain in the legs and difficulty breathing. Other symptoms may include weakness, headaches, or a burning, tingling or prickling sensation in the skin. Some people have episodes of severe pain, redness, and swelling (especially in the hands and feet).
A number sign (#) is used with this entry because thrombocythemia-3 (THCYT3) is caused by heterozygous germline or somatic mutation in the JAK2 gene (147796) on chromosome 9p24. Description Thrombocythemia-3 is an autosomal dominant hematologic disorder characterized by increased platelet production resulting in increased numbers of circulating platelets. Thrombocythemia can be associated with thrombotic episodes, such as cerebrovascular events or myocardial infarction (summary by Mead et al., 2012). For a discussion of genetic heterogeneity of thrombocythemia, see THCYT1 (187950). Clinical Features Mead et al. (2012) reported a 3-generation family with autosomal dominant inheritance of thrombocythemia.
Differential diagnosis Differential diagnoses include the other myeloproliferative neoplasms (polycythemia vera, primary myelofibrosis, chronic myeloid leukemia; see these terms), myeloid malignancies (myelodysplastic syndrome), causes of secondary thrombocytosis (inflammation, cancer, iron deficiency, asplenia) and primary familial thrombocytosis (see this term).
A number sign (#) is used with this entry because thrombocythemia-2 (THCYT2) is caused by heterozygous germline or somatic mutation in the MPL gene (159530) on chromosome 1p34. For a general phenotypic description and a discussion of genetic heterogeneity of thrombocythemia, see THCYT1 (187950). Clinical Features Ding et al. (2004) reported a 3-generation Japanese family in which 8 of 16 members had thrombocythemia, with a platelet count more than 600 x 109/L. Bone marrow biopsies were normocellular and normoplastic, except for increased megakaryocytes. Inheritance The transmission pattern of thrombocythemia in the family reported by Ding et al. (2004) was consistent with autosomal dominant inheritance.
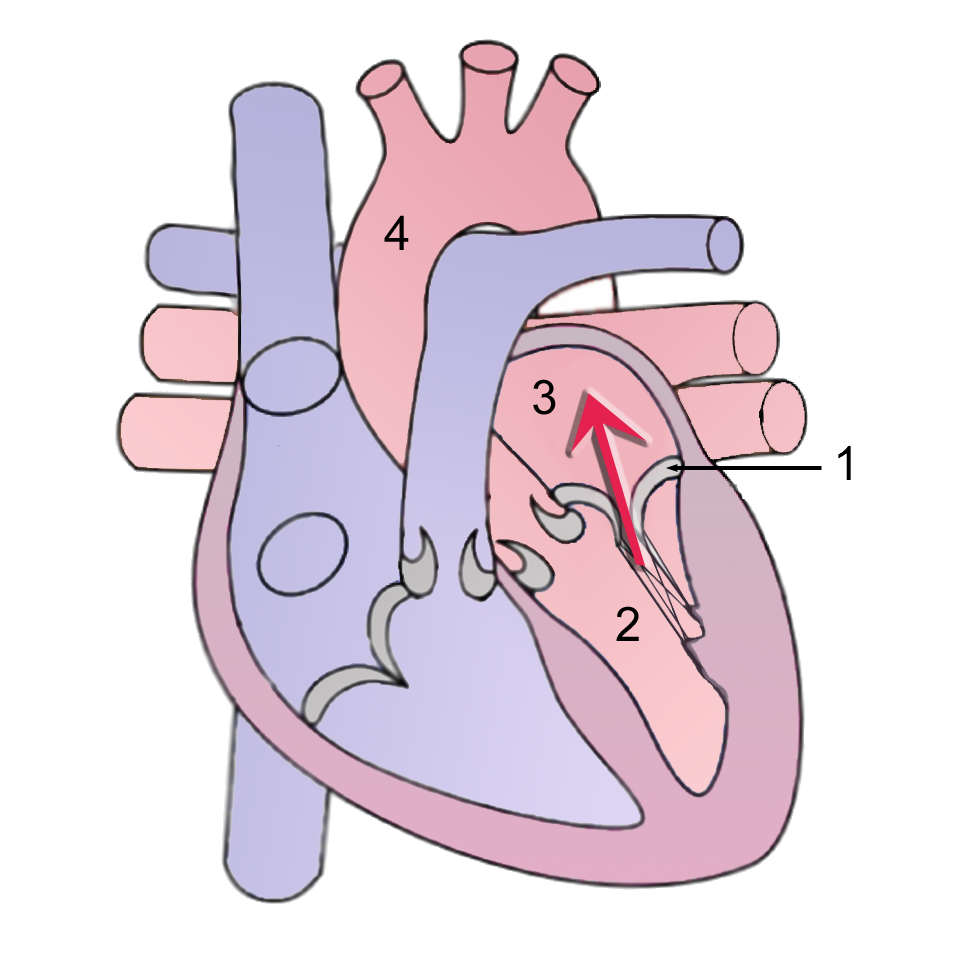
Overview Mitral valve regurgitation is a type of heart valve disease in which the valve between the left heart chambers doesn't close completely, allowing blood to leak backward across the valve. It is the most common type of heart valve disease (valvular heart disease). If the leakage is severe, not enough blood will move through the heart or to the rest of the body. As a result, mitral valve regurgitation can make you feel very tired (fatigued) or short of breath. Other names for mitral valve regurgitation are: Mitral regurgitation (MR) Mitral insufficiency Mitral incompetence Treatment of mitral valve regurgitation may include regular monitoring, medications or surgery.
Acquired Horner syndrome may result after trauma, neoplastic insult, or even vascular disease . ... Patients with third nerve palsy tend to have diminished or absent function of the levator. When caused by Horner's syndrome , ptosis is usually accompanied by miosis and anhidrosis . ... After diagnosing the types of ptosis the ophthalmologist will then decide if the patient is a good candidate for surgery. [11] Classification [ edit ] Mild right eyelid ptosis Depending upon the cause it can be classified into: Neurogenic ptosis which includes oculomotor nerve palsy , Horner's syndrome , Marcus Gunn jaw winking syndrome , third cranial nerve misdirection. Myogenic ptosis which includes oculopharyngeal muscular dystrophy , myasthenia gravis , myotonic dystrophy , ocular myopathy, simple congenital ptosis, blepharophimosis syndrome. Aponeurotic ptosis which may be involutional or post-operative. ... M.; Theakston, R. D. (1996). "The emerging syndrome of envenoming by the New Guinea small-eyed snake Micropechis ikaheka " .
This is called compensatory anti-inflammatory response syndrome (CARS). [11] Both the inflammatory and anti-inflammatory reactions are responsible for the course of sepsis and are described as MARS (Mixed Antagonist Response Syndrome). ... Septic shock may be regarded as a stage of SIRS (Systemic Inflammatory Response Syndrome), in which sepsis, severe sepsis and multiple organ dysfunction syndrome (MODS) represent different stages of a pathophysiological process. ... In rough order of increasing severity these are, bacteremia or fungemia; sepsis, severe sepsis or sepsis syndrome; septic shock, refractory septic shock, multiple organ dysfunction syndrome, and death. 35% of septic shock cases derive from urinary tract infections , 15% from the respiratory tract, 15% from skin catheters (such as IVs ), and more than 30% of all cases are idiopathic in origin. ... It is significantly greater when sepsis is left untreated for more than seven days. [34] References [ edit ] ^ "Acute respiratory distress syndrome: MedlinePlus Medical Encyclopedia" . ... "Compensatory anti-inflammatory response syndrome". Thromb. Haemost . 101 (1): 36–47. doi : 10.1160/TH08-07-0421 .
Other names for this type are complete or true precocious puberty. [4] Causes of central precocious puberty can include: damage to the inhibitory system of the brain (due to infection , trauma , or irradiation ) hypothalamic hamartoma produces pulsatile gonadotropin-releasing hormone (GnRH) Langerhans cell histiocytosis McCune–Albright syndrome Central precocious puberty can also be caused by brain tumors , infection (most commonly tuberculous meningitis , especially in developing countries), trauma, hydrocephalus , and Angelman syndrome . [5] Precocious puberty is associated with advancement in bone age, which leads to early fusion of epiphyses, thus resulting in reduced final height and short stature. [6] Adrenocortical oncocytomas are rare with mostly benign and nonfunctioning tumors. ... As an example, children with a very rare genetic condition called aromatase excess syndrome- in which exceptionally high circulating levels of estrogen are present- usually develop precocious puberty. Males and females are hyper-feminized by the syndrome. [14] The "opposite" case would be the hyper-masculinisation of both male and female patients with congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency , in which there is an excess of androgens. ... It is located on human chromosome 15 on the long arm in the Prader-Willi syndrome critical region2, and has since been identified as a cause of premature sexual development or CPP. [30] The identification of mutations in MKRN3 leading to sporadic cases of CPP has been a significant contribution to better understanding the mechanism of puberty. [31] MKRN3 appears to act as a "brake" on the central hypothalamic-pituitary access. ... Retrieved on 2009-02-20 External links [ edit ] Classification D ICD - 10 : E30.1 , E22.8 ICD - 9-CM : 259.1 OMIM : 176400 MeSH : D011629 DiseasesDB : 10519 External resources MedlinePlus : 001168 eMedicine : ped/1882 v t e Gonadal disorder Ovarian Polycystic ovary syndrome Premature ovarian failure Estrogen insensitivity syndrome Hyperthecosis Testicular Enzymatic 5α-reductase deficiency 17β-hydroxysteroid dehydrogenase deficiency aromatase excess syndrome Androgen receptor Androgen insensitivity syndrome Familial male-limited precocious puberty Partial androgen insensitivity syndrome Other Sertoli cell-only syndrome General Hypogonadism Delayed puberty Hypergonadism Precocious puberty Hypoandrogenism Hypoestrogenism Hyperandrogenism Hyperestrogenism Postorgasmic illness syndrome Cytochrome P450 oxidoreductase deficiency Cytochrome b5 deficiency Androgen-dependent condition Aromatase deficiency Complete androgen insensitivity syndrome Mild androgen insensitivity syndrome Hypergonadotropic hypogonadism Hypogonadotropic hypogonadism Fertile eunuch syndrome Estrogen-dependent condition Premature thelarche Gonadotropin insensitivity Hypergonadotropic hypergonadism
This condition is called McCune-Albright syndrome. A group of genetic issues, called congenital adrenal hyperplasia, that involve the adrenal gland making atypical hormones. ... This condition is called McCune-Albright syndrome. Being exposed to creams or ointments that contain estrogen or testosterone.
It may be caused by use of certain medications or supplements, infection, or any condition that leads to high levels of calcium in the blood or urine including hyperparathyroidism , renal tubular acidosis , Alport syndrome , Bartter syndrome , and a variety of other conditions.
These include renal colic , polyuria and polydipsia : [4] Renal colic is usually caused by pre-existing nephrolithiasis , as may occur in patients with chronic hypercalciuria . [4] Less commonly, it can result from calcified bodies moving into the calyceal system. [4] Nocturia, polyuria, and polydipsia from reduced urinary concentrating capacity (i.e. nephrogenic diabetes insipidus ) as can be seen in hypercalcemia, medullary nephrocalcinosis of any cause, or in children with Bartter syndrome in whom essential tubular salt reabsorption is compromised. [4] There are several causes of nephrocalcinosis that are typically acute and present only with kidney failure . [4] These include tumor lysis syndrome , acute phosphate nephropathy , and occasional cases of enteric hyperoxaluria . [4] Cause [ edit ] Nephrocalcinosis is connected with conditions that cause hypercalcemia, hyperphosphatemia, and the increased excretion of calcium, phosphate, and/or oxalate in the urine. ... In conjunction with Nephrocalcinosis, hypercalcemia and hypercalciuria the following can occur: Primary hyperparathyroidism : Nephrocalcinosis is one of the most common symptoms of primary hyperparathyroidism . [5] Sarcoidosis : Nephrocalcinosis is one of the most common symptoms. [6] Vitamin D : This can cause nephrocalcinosis because of Vitamin D therapy because it increases the absorption of ingested calcium and bone resorption, resulting in hypercalcemia and hypercalciuria . [1] Medullary nephrocalcinosis [ edit ] Medullary nephrocalcinosis in Sonography Medullary sponge kidney [7] Distal renal tubular acidosis [7] Hyperoxaluria [7] Renal papillary necrosis And other causes of hypercalcemia (and thus hypercalciuria ) Immobilization (leading to hypercalcemia and hypercalciuria) Milk-alkali syndrome Hypervitaminosis D [7] Multiple myeloma Hypercalciuria without hypercalcemia [ edit ] These conditions can cause nephrocalcinosis in association with hypercalciuria without hypercalcemia: Distal renal tubular acidosis Medullary sponge kidney Neonatal nephrocalcinosis and loop diuretics Inherited tubulopathies Chronic hypokalemia Beta thalassemia Mechanism [ edit ] Nephrocalcinosis is caused by an increase in the urinary excretion of calcium, phosphate, and/or oxalate. [1] Nephrocalcinosis is closely associated with nephrolithiasis , and patients frequently present with both conditions, however there have been cases where one occurs without the other. [1] Calcium oxalate and calcium phosphate crystals form when the concentration of the reactants exceeds the limit. ... External links [ edit ] Classification D ICD - 10 : E83.5 † N29.8 * ICD - 9-CM : 275.49 MeSH : D009397 DiseasesDB : 8902 External resources MedlinePlus : 000492 eMedicine : article/243911 Patient UK : Nephrocalcinosis v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
IRF6-related disorders include two different disorders caused by abnormalities in the interferon regulatory factor 6 (IRF6) gene. Van der Woude syndrome (VWS) is at the mild end of the spectrum and popliteal pterygium syndrome (PPS) is at the severe end of the spectrum.
Disturbance of ridge formation resulting in scattered short ridges or in ridges simply comprised of irregular dots is a feature in patients with Down syndrome and in some patients with limb malformations. ... It would be of interest to study cultured epidermal cells from persons with this disorder and persons with absence of fingerprints (136000). Also see Basan syndrome (129200). Inheritance - Autosomal dominant Skin - Patternless dermal ridges ▲ Close
Jejunal atresia (243600) occurs in several forms, one of which type IIIb ('apple peel' syndrome), most commonly shows familial occurrence. ... The authors suggested that this association of abnormalities is a previously undescribed syndrome with autosomal dominant inheritance.
Reif-Lehrer (1976) reported that 25% of persons develop the 'Chinese restaurant syndrome' on exposure to monosodium glutamate (MSG) used as a flavor enhancer. ... Symptoms of the Chinese restaurant syndrome include tightness in the back of the neck, pressure behind the eyes, frontal or temporal headache, facial flushing, nausea, etc.
Teebi et al. (1998) concluded that this constellation of features could represent a previously unrecognized syndrome either in the category of syndromic hereditary spastic paraplegia or XY sex reversal.
Find sources: "Malnutrition–inflammation complex" – news · newspapers · books · scholar · JSTOR ( August 2008 ) ( Learn how and when to remove this template message ) Malnutrition–inflammation complex Differential diagnosis chronic heart failure Malnutrition–inflammation complex (syndrome), abbreviated as " MICS " and also known as " malnutrition – inflammation – cachexia syndrome ", is a common condition in chronic disease states such as chronic kidney disease (where it is also known as uremic malnutrition or protein–energy malnutrition ) and chronic heart failure .
X-linked immunoneurologic disorder is characterized by immune deficiency and neurological disorders in females, and by neonatal death in males. Epidemiology The syndrome has been described in only one family with nine affected individuals (five males and four females) spanning two generations. ... Etiology The gene locus has been mapped to Xq26-qter. Differential diagnosis The syndrome should be considered in the differential diagnosis of hereditary spastic paraplegia in females and of other causes of severe neonatal hypotonia in males.